While healthcare providers look to the future generation of biosimilars as a means of reducing costs, Marie Rock, Ph.D, vice president, protein bioanalysis group, Midwest BioResearch, a subsidiary of WIL Research, reviews some important issues raised and calls for side-by-side clinical trials for biosimilars approval.
Word has spread that biosimilar regulation in the US might have a more concrete pathway to follow in the coming months. According to United States Food and Drug Administration (US FDA) Commissioner Margaret Hamburg, the FDA plans to unveil rules for reviewing the first copies of biosimilars ‘very soon’. This news comes a year after the passing of the Patient Protection and Affordable Care Act, the first piece of regulatory legislation for biosimilars in the US. To date, the Act has generated more questions than answers.
Passed in March 2010 by President Obama, the Act was designed to cut healthcare costs by creating a financially less intensive approval pathway for new biologics that are deemed ‘highly similar’ or biosimilar to its originator. Unfortunately, the Act fails to provide exactly how a ‘highly similar’ determination is made. As a result, two camps have risen out of this ambiguity – those in favour of full blown side-by-side clinical trials and those opposed to extensive clinical testing.
Supporters of side-by-side clinical trials of both the originator and the biosimilar argue that this approach better ensures patient safety. Opponents argue that extensive clinical testing may be unnecessary if a biosimilar is shown to be highly similar to a reference product’s published data – an approach that has proved extremely successful for rolling out cost-friendly generic small molecule drugs.
However, unlike small molecule drugs and their generics, it is impossible precisely to characterise biologics and biosimilars chemically and technically. As a result, the determination of a ‘highly similar’ assessment is based on the nature of the biologic and a possible harmful immunological response. For those working in the patients’ interest, these are factors that greatly outweigh any economic advantage.
Complex characteristics of biologics call for a thorough case-by-case set of clinical trials of both the originator and the biosimilar using current analytical methods and expectations – considerations that will hopefully be taken into account as the FDA introduces its new rules.
black and white for generics
The well-defined, predictable nature of small molecule drugs combined with the rigorous testing done by originators enables generic manufacturers to bypass several costly steps, as mapped out in the Drug Price Competition and Patent Term Restoration Act (Hatch-Waxman Act) passed in 1984.
Following Hatch-Waxman’s guidelines, generic drug companies are spared the costs of expensive clinical trials because all they need to prove is that their drug’s active ingredient is ‘bioequivalent’. One measure of a generic drug’s bioequivalence is its bioavailability – the amount of the drug in the bloodstream, the time it takes to get there, and the time it takes to exit the body. If the concentration and absorption of the generic meet statistical requirements compared with the originator, the generic is considered to be bioequivalent.
In addition, since the generic drug is the same molecule, all additional information regarding safety and metabolism has been proven and accepted by the FDA in the submission provided by the originator (Table 1).
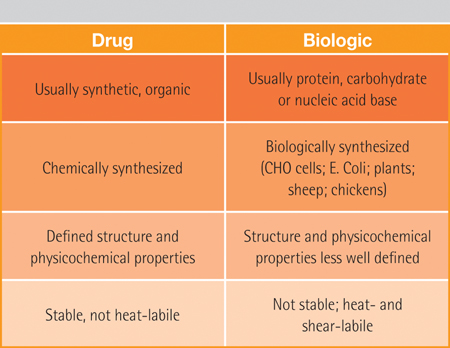
Table 1: A comparison of the origin and production of traditional small molecule drugs and typical biologics
Opponents of side-by-side clinical trials have taken the Hatch-Waxman Act as a model for biosimilar approval, hoping that such an amended approval process will reduce healthcare costs. Those looking at biosimilar regulation through economically motivated lenses fail to realise that to ensure patient safety and efficacy, side-by-side clinical trials are needed.
grey for biosimilars
Unlike small molecule drugs, biologic drugs are large complex molecules and scientific characterisation of biologics doesn’t produce absolute, well-defined metrics. Variables such as protein folding, aggregate formation and glycosylation can affect the performance and efficacy of the biologic. Choices made during production can also influence the nature of the biologic, including the choice of the cell type, development of the genetically modified cell, purification process and formulation of the therapeutic protein.
Because of these complexities, originator companies still have problems replicating their own production process despite years of experience with the drug under patent protection. Furthermore, these complexities make exact replication of the originator’s active molecule nearly impossible (Table 2).

Table 2: The different nature of the two types of drugs
The analytical methods used to test biological drugs are constantly improving in the hopes of better quantifying their complex characteristics. Many of these techniques and technologies were not available during the development of the originator. Data published on originators against which opponents of full-blown clinical trials would like biosimilar manufacturers to compare their drug are outdated.
As a result, thorough side-by-side clinical trials of the originator and the biosimilar are needed to provide analytical evidence of bio-similarity. Two of the most important and most difficult aspects of biosimilar clinical testing are providing evidence of bioequivalence (purity and potency) and immunogenicity (safety).
As with generics, the bioequivalence of biosimilars must be tested, but the complexity of biological drugs necessitates side-by-side clinical trials. The bioequivalence of biosimilars must be established through assays using antibodies to ‘extract’ the biologic from the sample. Using a biologically derived technique to assess a biologically-derived drug further complicates testing.
bioequivalence
Biologic drug assays are highly diverse and results vary from test to test. For example, an assay that shows similar profiles between an originator and biosimilar does not necessarily indicate that the two are bioequivalent. If the antibody binds to the same molecular component in both the originator and the biosimilar, although it may be ‘seeing’ the drugs in the same way, differences may still exist in the other parts of the drug.
On the other hand, an antibody may bind differently to an originator and biosimilar due to their unique glycosylation patterns. Though they may be bioequivalent, they cannot be considered biosimilar because the antibodies ‘see’ them differently. These differences may affect the effectiveness of biosimilars and the way the body reacts to them.
Immunogenicity tests for generics are not necessary because small molecule drugs do not typically cause an immunogenic response. With biological drugs, the slightest presence of any impurities, degraded protein, or aggregates can trigger an antibody response. An anti-drug antibody response can lead to a range of outcomes from neutralisation of the drug’s therapeutic action to more serious consequences such as a cross-reaction with an endogenous protein.
side by side tests
As another challenge for biosimilars, methods currently required for immunogenicity testing must meet more rigorous performance expectations than those that were in effect for the originator. For example, if the originator reported an immune response of 2% then, and if the same set of samples were tested on methods meeting current standards, a rate of 5–10% may have to be reported now.
As a result, side-by-side testing is necessary for comparing rates of immunogenicity; otherwise, the follow-on may paradoxically appear to have a greater immune potential.
In addition to bioequivalence and immunogencity, the FDA must also consider how much drift can be tolerated, a characteristic unique to biologics. Unlike small-molecule drugs, the slightest change in the manufacturing process can lead to slight changes in the final product. The complex nature of biologics makes it almost impossible to make identical copies of the drug. These changes can lead to unexpected changes in patient response in terms of safety and efficacy.
For originators and biosimilars alike, it is not uncommon for a biologic drug to differ slightly from batch to batch. With this in mind, the FDA must decide the degree to which the biosimilar must drift along with the originator. In other words, should the biosimilar be referenced against the drifted reference product or the original reference product? If compared with the original reference product, the biosimilar may be referenced to a product that is no longer being made by the originator, i.e. no longer exists. However, to require biosimilars to drift along with the originator would be a difficult and costly process.
Providing patients with generic versions of name brand drugs has proved to be successful in cutting healthcare costs for small molecule drugs. However, the complex nature of the biologics production process and the level of testing that should be required of such unpredictable drugs eliminate the economic advantage of producing a biosimilar.
Biosimilars, for the most part, won’t be able to provide the dramatic cost reduction patients are used to seeing in small molecule drugs. But, that doesn’t mean that biosimilars are without their benefits. Innovation of biosimilars should continue because it is precisely their complex nature that makes it so difficult to establish biosimilarity that also makes them valuable.
Patients taking biologics may begin to see decreased therapeutic efficacy as their immune system begins to adjust by producing antibodies against the biologic or the related impurities. Patients who become antibody positive to the originator have an option for an alternative therapy provided by the biosimilar (Figure 1). Since the biosimilar is not likely to be identical to the originator, there is a chance that the antibodies to the originator will not interfere with the efficacy of the biosimilar.
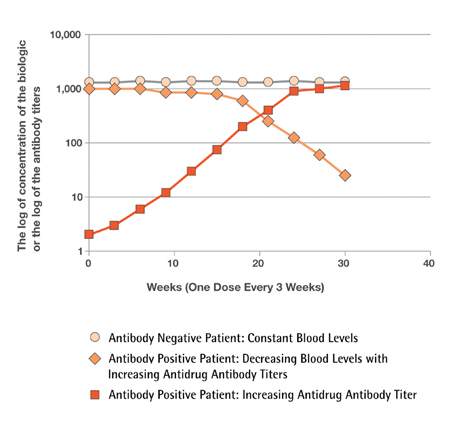
Figure 1
Figure 1 shows circulating blood levels of a biologic in two patients tested every three weeks for a period of 30 weeks. One patient is negative for anti-drug antibodies (round symbols) and the other patient is developing an anti-drug antibody reaction (orange diamonds) during the course of the treatment. The antibody negative patient (round symbols) shows constant blood levels for the whole time and benefits from the therapy. The antibody positive patient shows a rise in the anti-drug antibody reaction (red squares). As the antibody reaction progresses, there is a decrease in the blood levels (orange diamonds) as a result of antibody interactions with the biologic. This particular immune response results in the biologic’s loss of efficacy, and symptoms return. This is where an alternative choice, i.e. a biosimilar, can be critical to the antibody positive patient population