The need for greener chemical manufacturing processes is driving research into the discovery of catalysts that provide greater selectivities using both benign reagents and non-hazardous reaction conditions. Dr Sarah Houlton reports on some recent API reaction successes.
Many structural features that are often present in APIs can prove challenging to synthesise, especially at the scale required for commercial production. While it may be possible to make them in the lab, in practice the route may involve conditions that are not conducive to scale-up, such as very high or low temperatures, or hazardous reagents. Alternative routes need to be developed, and often a metal catalyst can provide the answer to a synthetic problem by enabling a reaction to take place that would not otherwise be possible. They can be especially useful in installing stereochemistry into an otherwise achiral or racemic molecule.
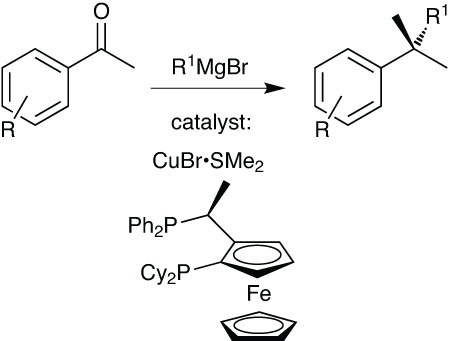
Chiral tertiary alcohols are a particular problem. While they are common structural features in APIs, they are difficult to make, and the synthetic tricks such as asymmetric hydrogenation followed by dynamic kinetic resolution that can work well for secondary alcohols simply do not apply to tertiary ones. Racemic tertiary alcohols are rarely amenable to resolution, and so the stereocentre must be constructed in a selective way.






