Testing for elemental impurities is a vital step in drug product manufacturing to ensure that the quality, purity and consistency of the pharmaceutical yield is maintained. Until recently, manufacturers were required to carry out a wet chemistry heavy metals test to meet the industry standards regulated by the United States Pharmacopoeia (USP) and the European Pharmacopoeia (EP).
Recent updates of these legal frameworks have removed the test to be replaced by specific elemental testing against validated methods using expensive analytical equipment.
The general approach now accepted for the control of elemental impurities is based on a risk assessment, evaluating the likelihood of contamination of target elements from the materials and processes used in the production of the final product, as opposed to enforcing mandatory testing for all elements in all products, which would have significant cost and time implications.
During the winter of 2017/2018 the requirement for the control of elemental impurities in drug products became mandatory within the USP and EP. These changes had long been proposed but faced a certain degree of resistance from the industry, particularly for USP compliance.
In December 2014 the International Council for Harmonisation (ICH) finalised its Q3D regulations1 for control of elemental impurities. This document contained a toxicological assessment of elemental impurities and used this information to establish permissible daily exposure limits (PDEs) on 24 elements of specific concern. It also provided advice for controlling these components and suggested a risk based approach for them.
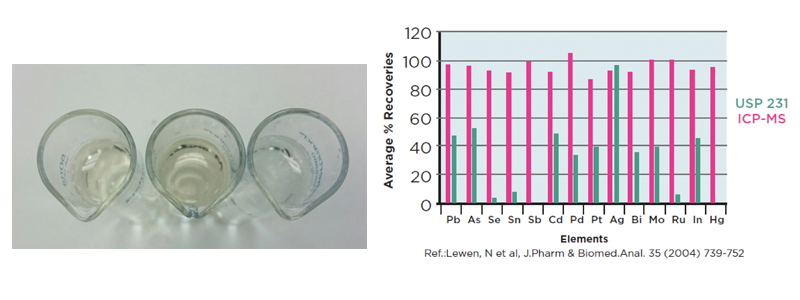
Figure 1: A visual comparison of the heavy metals test, the left hand tube is the sample, the right hand tube is the blank and the middle tube is a 10 ppm lead standard. Relative recoveries of the heavy metals elements by ICP-MS and the heavy metals test from the USP
These guidelines were accepted by both the USP and EP, and in terms of limits, these have now been coordinated across these pharmacopeias. These changes have now been in place for nearly six months, so are still in the early stages of being part of the regulations. It has, however, had a significant impact on the nature of pharmacopeial testing.
There are several reasons why the USP chose to eliminate the simple heavy metal test, including: the test lacked sensitivity to determine the low levels of heavy metals now expected in industry standards; it lacked specificity – it did not have the ability to distinguish between and quantitate heavy metals of concern; and the test used highly toxic reagents.
Given the scale of the impact of these changes, one may question why they were brought into the pharmacopeias? The answer to this is simple: the existing testing was no longer fit for purpose and bore no reflection on any toxicological information, rather the limits were set by the performance of the test itself.
The heavy metals test explained
The obsolete heavy metals method was based on wet chemistry test, which first appeared in the USP in 1905 and utilised a colour change of a preparation of the product compared to a known reference standard. This method had several problems: it was non-specific, prone to low recoveries and very subjective; the colour change was quite subtle and reliant on the operator; it was also troublesome if a coloured test solution was obtained. Studies carried out have also shown that some elements gave poor recoveries – as low as 2% for mercury – most likely due to the high temperatures used in the preparation of the samples.
As a result of these limitations, along with improved toxicological information and access to more powerful, user-friendly and cheaper analytical equipment, it has been suggested that this method should be replaced by more specific instrumental techniques, thus allowing targeted analysis of individual elements, in a quantitative manner, enabling sensible scrutiny of the potential toxicity of the materials from the presence of toxic metal components.
These changes were worked on independently by the USP and EP, with the USP concentrating on the toxic elements such as mercury and lead, based on the potential to cause harm, whereas the EP focused more on catalytic residues, such as palladium and platinum, as these were more likely to be present, if less toxic than the heavy metals.
At the same time, the ICH was also working on a project to look at elemental impurities, this was finalised in December 2014. By this point, the USP and EP had their own proposed regulatory approach. On the basis of work from the ICH, and after much discussion and changes of dates, both pharmacopeias agreed to incorporate the ICH guidance.
The ICH guidelines
The guidance issued by the ICH includes 24 elements and grouped into four categories based on their relative toxicity, likelihood of occurrence, and route of administration. These elements (see table 1) have been assigned exposure limits based on the toxicology and the route of administration. The guidance aims to assist manufacturers in adequately establishing the risk of contamination in the final products, which therefore poses a risk to patient safety.
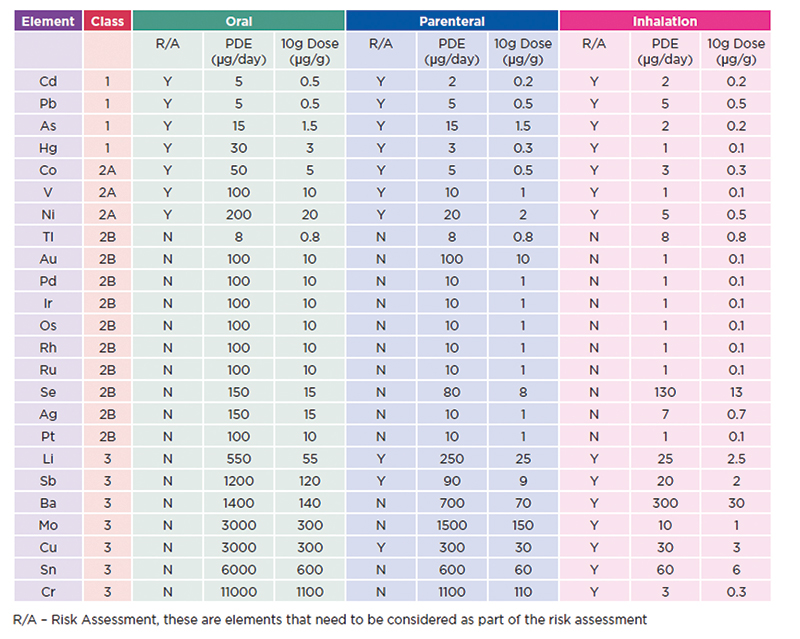
Table 1: ICH elements and limits These values are given as permissible daily exposure limits (PDE in µg), but also the permitted concentration in drug products are included (as µg/g), using the assumption that the maximum dosage will be 10 g per day
The specific toxicological profile of each element, dependent on its class and route of administration, is used to highlight when an element is or is not required for consideration as part of the risk assessment, allowing the process to be simplified.
If an element has been used in the manufacture of – or added to – the pharmaceutical product then it must be included as part of the risk assessment. If an element is not intentionally added then its inclusion as part of the assessment is based on the toxicity of the material, the likelihood of being present and the route of administration, with this information set out in the Q3D regulations guidance.
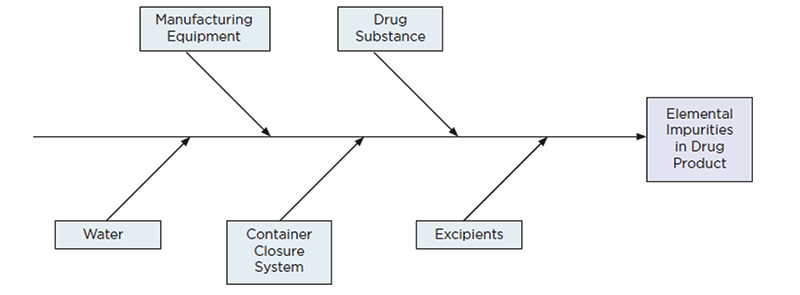
Figure 2: The manufacturing process should also be considered as part of the risk assessment and a fish-bone diagram is commonly used to ensure that all aspects of the process are considered
Risk assessment under ICH guidelines
Under the new USP testing standard, the focus is on finished products being tested in their marketed containers with the labelling in place. The emphasis on risk assessment is encouraged to evaluate the likelihood of target element contamination of the materials and processes used in production of the final product. This is in contrast to a mandatory test for all elements in all products being enforced.
Elements deliberately added to the process are automatically included in the risk assessment (mainly as catalysts seen in class 2B). The class 1 (most toxic) and class 2A elements (most likely to be present as contaminants) should also be included in the risk assessment, class 3 are typically only considered for non-oral dosage forms.
In general, each of these factors has its own level of risk for introduction of elemental contamination. Water, for example, is probably of low risk if generated under GMP conditions. The manufacturing equipment may pose a risk of the addition of class 2A elements, from wear and contact of steel materials used in the production line, but with relatively high limits for these elements along with robust controls and cleaning protocols the impact should be minimal.
Packaging, in general, poses a low risk, and information from extractable and leachable studies may provide useful information for the risk assessment. The drug substance is the most likely route of contamination from class 2B as residual catalyst materials, but this risk could be relatively low, particularly if the catalyst is used early on in the synthesis and is removed through purification steps.
It is generally accepted that the highest risk of contamination comes from the excipients used and particularly the use of mined excipients as these are typically mineral products likely to contain elements of concern, which may be tricky to eliminate as part of the refining process.
Once the risk assessment has been completed, it may be decided the risk of contamination is sufficiently low that no testing is required, but in many cases there may be potential for contamination.
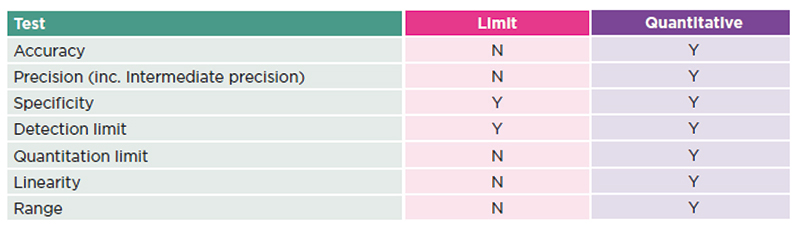
Table 2: ICH validation parameters required
Contamination risk for materials
The ICH Q3D document states that by establishing the elements of concern are not detected at 30% of the established PDE for the specification in a certain number of batches of material, e.g. three production batches, then the potential of contamination above the specified limits is deemed to be sufficiently low that potentially no further controls are required.
An elemental screen is useful to obtain information about the levels of elements to establish whether this limit is exceeded. This is a generic method of analysis utilising inductively coupled plasma – mass spectrometry (ICP-MS), a powerful multi-element technique that enables the analysis of most elements down to sub parts per billion (ppb) levels and gives an overview of the elemental composition of the sample. If the screen does indicate the levels of elemental contamination exceeds the 30% threshold then it is likely a more targeted analysis will be required. This would therefore require a method of validation to be carried out in accordance with the relevant pharmacopeias.
There is the flexibility to carry out different types of validation. For products that are solely for the US market, Chapter <233> sets out validation parameters and methodology for compliance with the limits described in Chapter <232>. For other markets, a standard validation according to the parameters set out in the ICH Q2 documents would be a useful approach. As this is a limit test, it is possible that a simplified validation could be carried out, but this would only provide a discrete, pass or fail result.
It is often better to perform a fully quantitative validation as this will allow for numerical results to be obtained, which could in time provide sufficient evidence to reduce the burden of testing, even potentially demonstrating that the risk of contamination is sufficiently low so that testing is no longer required.
This article first appeared in the June issue of Cleanroom Technology.