It is therefore critical to have a unified understanding of all relevant definitions, as described below.
- PDE (Permitted Daily Exposure), also known as ADE (Acceptable Daily Exposure), represents a substance-specific dose that is unlikely to cause an adverse effect if an individual is exposed at or below this dose every day for a lifetime.
- NOEL (No Observed Effect Level) is the highest tested dose at which no “critical” effect is observed. If the critical effect is observed in several animal studies, the associated NOEL should be used to calculate the PDE value.
- OEL (Occupational Exposure Limit) is the average concentration load of an airborne active pharmaceutical ingredient (API) in µg/m3 that’s acceptable during an 8-hour time-weighted average with no negative impact on workers/environment.
- OEB (Occupational Exposure Band) is the banding of OEL into ranges for which appropriate measures to protect workers and facilities/equipment can be defined in common for APIs with different OELs within this band.
- COSHH (Control of Substances Hazardous to Health) assessment.
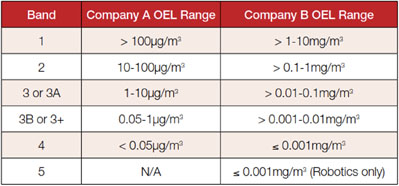
Table I illustrates the difference in banding applied by two pharmaceutical companies, demonstrating that there is a significant lack of common terminology and, therefore, a true understanding of potent classification.
When developing and manufacturing highly potent drug products, there should always be an OEL monitoring programme at each stage of the process. Strict cross-contamination controls are essential and a risk assessment should be done according to four principle modes:
- airborne
- mechanical
- personnel transfer
- retention of product on contact surfaces.
With traditional non-potent drug products, avoiding contamination from personnel involved in the production process is, of course, critical. However, when manufacturing drug products containing highly potent APIs (HPAPIs), operating in rooms under negative pressure isn’t enough.
Table I: Inconsistent definitions of OEL bands are common throughout the pharmaceutical industry
High-grade specialised containment equipment is also required to protect employees from the API itself — and to ensure drug product integrity in a multiproduct facility.
Primary and secondary containment
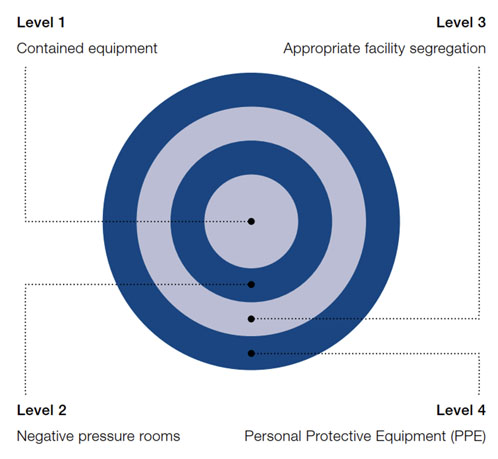
Understanding the levels of containment is important when determining the appropriate handling of highly potent compounds (Figure 1). Designing a safe and compliant high potent containment facility is a demanding process.
Primary containment requires the use of suitable equipment that runs under negative pressure and effectively serves as a “cleanroom” in its own right, with secondary containment being the facility itself.
The appropriate solution depends on the product potency and batch size being handled, as different solutions may need to be considered for laboratory, small- and commercial-scale use.
Figure 1: Levels of containment
Containment must therefore address the potency of the product with careful consideration being given to dust extraction (heating, ventilation and air-conditioning systems [HVAC] and central dust collection) and breach control procedures.
HVAC systems should be controlled using a building management system (BMS), which must be suitably designed with terminal high efficiency particulate air (HEPA) filters (H14 grade). Additional safeguarding measures such as single-pass air ensures a full room evaluation of any potential loss of containment, suitable automatic equipment cleaning and waste disposal systems.
In addition to the room pressure differentials, airlocks adjacent to the manufacturing area are necessary for high risk activities such as API dispensing, along with utilities such as purified water and effluent treatment capabilities.
Restricted access is also a requirement, ensuring that only essential staff can access the HPAPI processing areas, with cleaning and decontamination areas provided for employees.
Containment via QbD
A strategy based on Quality by Design (QbD) is necessary to ensure process reliability and reproducible product quality, with scientifically designed products and processes and the control of critical process parameters (CPPs), critical quality attributes (CQAs) and the quality target product profile (QTPP).
As part of the design, modern process analytical technology (PAT) tools are desirable for inline real-time data collection as the equipment cannot be opened during processing. To eliminate manual intervention, automatic adjustments of any manufacturing processing parameters should be done using PLC-controlled equipment with data trending.
Statistical packages and experimental design assessment tools should also be employed to reduce development times and the costs associated with drug losses and unnecessary manufacturing. A focus on continuous process monitoring and process verification during routine manufacturing provides
- reliable product quality outcome
- increased throughput
- high planning precision
- low waste and inventory costs
- increased operator safety.
New product introduction
When handling potent materials, new product introduction (NPI) is a key consideration. An industry leading CDMO should have robust processes in place to assess all new molecules for their OEL and their PDE — prior to being accepted on site.
For example, a COSHH assessment process would define the molecule, its mode of action and the appropriate handling requirements, maintaining operator and environmental safety at all times.
Toxicological and pharmacological assessments of all molecules is a requirement, both to protect the scientists and operators, and to provide a suitable cleaning assessment and cleaning verification parameters to eliminate the risk of cross-contamination to the next product.
A good manufacturing practice (GMP) Failure Mode and Effect Analysis (FMEA) assessment is also generated, supporting data from the safety, licencing equipment and premises data review to ensure the product can be safely processed.
Specific to technical transfer projects, Excipient Gap and Process Equipment Gap analyses are done to provide a summary of the site capabilities — aligned with the transfer site — and highlight any gaps that must be mitigated prior to initiation.
Only after all reviews and assessments are complete will a molecule be issued with its COSHH assessment for handling within the facility.
Cleaning philosophy
Each new molecule entering a multiproduct facility adds further complexity, requiring a robust cleaning philosophy.
Starting at NPI, the process should include methods to test and detect detergents and drug substances with swab and rinse samples and any additional cleaning that may be required. Equipment capabilities should be constantly reviewed, with oversight of all cleaning verification and validation, resulting in a science- and risk-based approach to the prevention of cross-contamination.

Employees should be provided with general GMP personal protective equipment (PPE), such as laboratory coats/suits and footwear, hairnets, eyewear, face masks and gloves, whereas powered air purifying respirators (PAPR) should be considered as part of a breach control and system failure programme.
But PPE should never be considered as part of the primary protection barrier during potency assessment or normal operations. Operator safety is of critical importance, resulting in a new way of thinking to develop drug products in a controlled facility with programmable logic controller (PLC) equipment.
Summary
As the market continues to evolve with increasing numbers of highly potent molecules in development, it is important to understand the requirements for the safe processing of such molecules, the differing regulatory requirements across the world and — above all — the safety of employees and the environment.
Sponsors with drug products containing HPAPIs often recognise that they have limited in-house capacity or experience. This leads them to outsource their development and manufacturing activities to a CDMO with established capabilities in the handling of such materials and a history of complying with applicable regulations.
Cost constraints and risk mitigation are other factors that may influence the decision to outsource. For pharmaceutical developers looking to contract out the development and manufacture of products containing HPAPIs, the challenge is finding a CDMO with the right technical capabilities, knowledge and experience to successfully manage the project.
Choosing a contract development and manufacturing organisation to develop and manufacture highly potent drug products is a complex process, requiring a high level of due diligence.
Assessing a CDMO’s capabilities and experience in the handling of potent drug substances, their regulatory expertise and compliance history, and their capacity to provide long-term support for clinical and commercial supply, is therefore central to the success of any outsourcing decision.
