As semisolid topical formulations become more prevalent, in vitro release testing (IVRT) is playing an increasingly vital role in determining the release performance and diffusion of these drug products — saving both time and cost compared with traditional bioequivalence studies. Neha D. Singh, Research Scientist, Formulation Development at Recipharm in Research Triangle Park, examines IVRT method development and looks at how the technique has evolved to optimise formulation processes and ensure manufacturing quality and consistency.
IVRT for semisolid topical formulations
Novel or complex dosage forms exhibit significant variability in formulation design, making it difficult to devise a single testing system that can be used to study the drug release properties of every dosage form. Different apparatus, procedures and techniques have been employed on a case-by-case basis, often specific to the dosage form category, formulation type and individual product characteristics.
Traditionally, the most commonly used quality control tests for topical dermatological preparations have included identification, assay, homogeneity, rheological properties, specific gravity and particle size determination; however, these tests provide little information about drug release properties or the effect of processing and manufacturing variables on the performance of the finished product.
As an effective research and development technique, IVRT can be used to develop and optimise formulations while also providing a quality control tool to assess manufacturing quality with time. The testing method measures the rate of release of a drug across a non-interactive membrane into an appropriate receiving medium. This process allows for the appropriate selection of a clinical candidate with Quality by Design (QbD) principles, and can serve as a cost-effective means to monitor product consistency.
FDA’s Non-Sterile Semisolid Dosage Forms for Scale-Up and Post Approval Changes (SUPAC-SS) guideline recommends IVRT for topical drug product testing, documenting that “in vitro release testing has shown promise as a means to comprehensively ensure consistent delivery of the active component(s).”1 The guidance recommends that IVRT be performed to compare equivalency between pre- and post-change batches. Recently, the industry has seen increasing demand for more reliable and reproducible IVRT methods, which calls for more exploration into how the technique can be further optimised.
Principle and theory behind IVRT
Human skin is a natural barrier to outside agents; consequently, drug products administered through semisolid formulations, such as creams, ointments and gels, must penetrate the skin to have an effect. An appropriate IVRT testing method needs to mimic skin permeation kinetics, including donor, membrane and a receptor medium that is analysed for drug concentration. Drug release from semisolid dosage forms follows the simplified Higuchi equation (Equation 1).2
Equation 1: Q = 2C0 • (Dt/n)1/2
Where
Q = amount of drug released per unit area of application
C0 = initial concentration of drug
D = diffusion coefficient of drug
t = time.
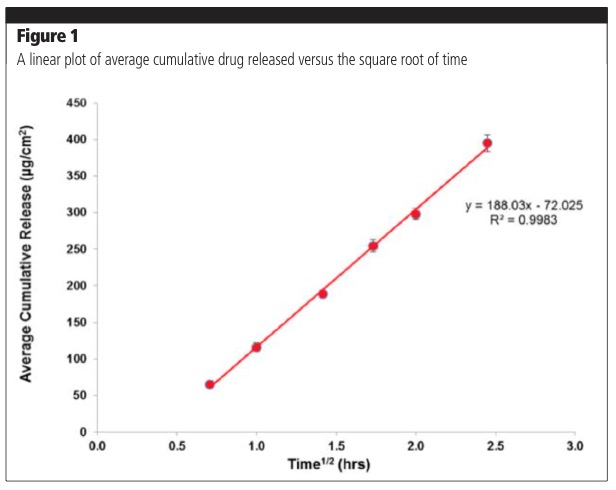
The equation is applicable when the drug is solubilised in a formation matrix and the amount of drug released at the end of the experiment is less than 30%. The amount of drug released is proportional to the square root of time; therefore, a plot of average cumulative release versus time should yield a straight line, the slope of which is used to calculate flux (amount released/cm2/h).1,2 The release of the drug is studied during a period of 4–6 hours, which is the typical duration of application for a topical product. Figure 1 demonstrates a typical release profile in line with Equation 1. Exceptions to this rule may be encountered with novel formulation approaches that have altered release profiles.
Best practices in IVRT method development
Successful IVRT is contingent on reliable drug transport from the test material through a membrane and into the receiving medium. Therefore, in identifying the optimal experimental parameters, the focus is on the active pharmaceutical ingredient’s physiochemical properties and selecting the proper membrane, receiving medium and sampling schedule.
IVRT method development involves a number of steps. Solubility screening is first used to develop a receptor medium that prevents saturation and maintains sink conditions. A membrane is then selected that has no leachables, minimises drug binding and has no rate limiting effect on release. This is followed by the selection of appropriate testing equipment, with the Franz diffusion cell assembly being the most commonly used apparatus to test semisolid topical dosage forms.
The formulation dose then needs to be determined, followed by the selection of time points to evaluate the release profile (SUPAC-SS advises a minimum of five time points). Other parameters such as sample volume, stirring speed, method of formulation addition and temperature of the receptor solution are also evaluated. Employing this process requires experienced analytical scientists that can develop and validate high-performance liquid chromatography (HPLC) or liquid chromatography-mass spectrometry (LC-MS) methods for high throughput sample analysis.
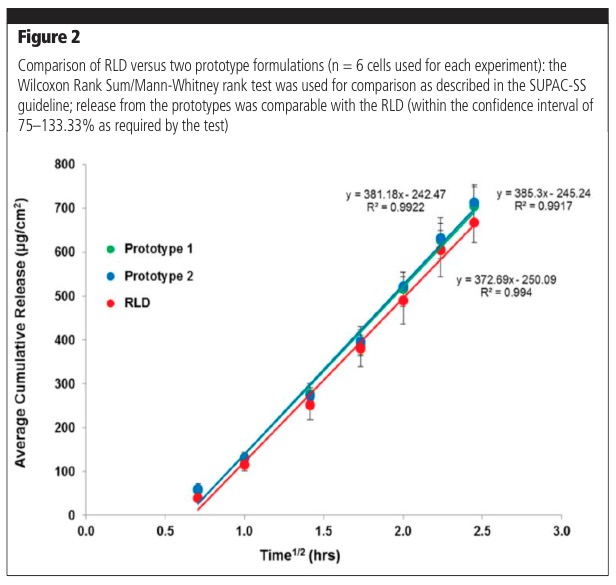
The IVRT methodology can be used to compare formulation prototypes to aid in product development, as well as compare generic formulation prototypes with a Reference Listed Drug (RLD) or a comparator. Figure 2 shows typical results for comparisons of prototype formulations with the RLD.
IVRT method validation
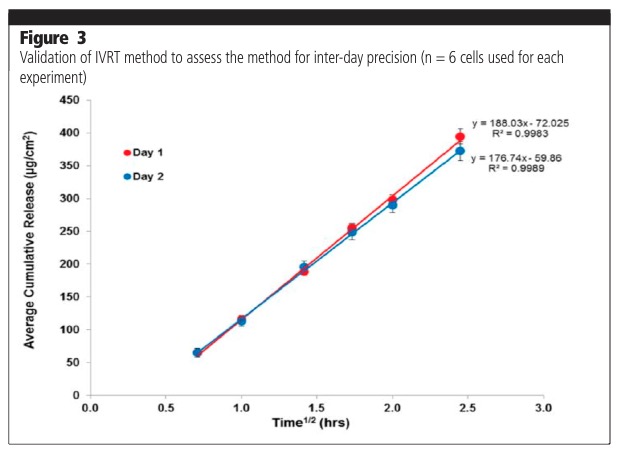
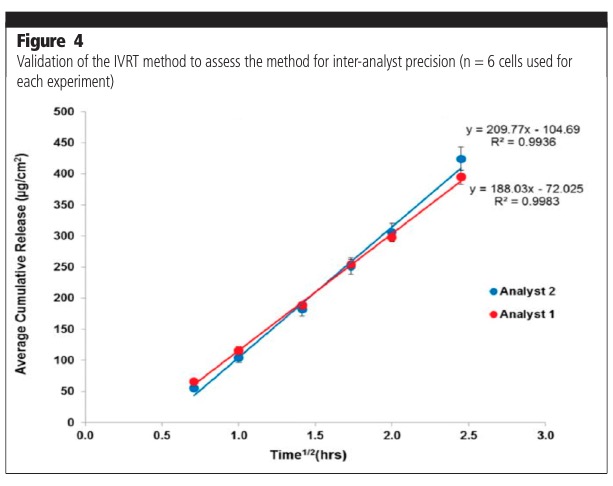
The parameters assessed as part of a method validation process consist of, but are not limited to, precision (repeatability, intermediate precision, reproducibility/ruggedness), robustness, method discrimination (ability of the method to differentiate between similar formulations) and mass balance at the end of the experiment. Figure 3 shows typical examples of intermediate precision analysis on two different days (day-day variability). Figure 4 shows analyses undertaken by two different analysts (analyst to analyst variability). The release profiles from the experiments were shown to not be significantly different.
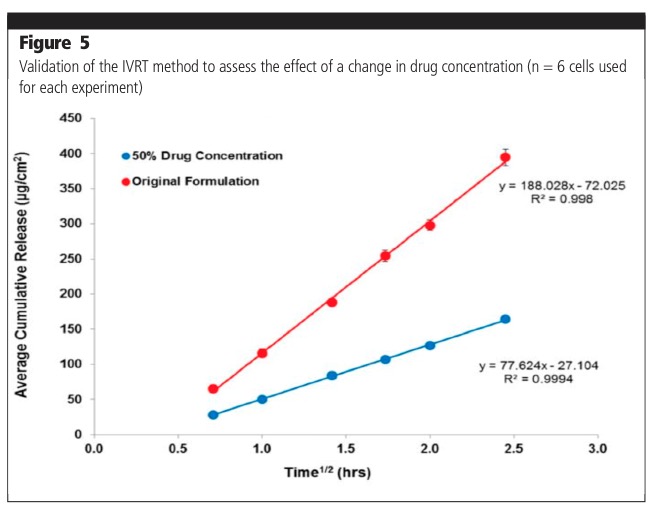
The IVRT method was also validated to assess the effect of change in drug concentration (Figure 5). Release from the original formulation (100% drug) was shown to be significantly higher when compared with a formulation containing 50% drug product. The effect of a change in formulation composition was also evaluated. The release from 50% excipient and 150% excipient formulations was shown to be significantly different compared with the original formulation.
Method benefits
The validated IVRT method provides an easy-to-use tool to evaluate the in vitro release profiles of topical drug products. The method can be used to ensure optimal thermodynamic activity and ensure that batch-to-batch performance and quality is uniform throughout a formulation’s shelf-life, which is essential for regulatory filings. The technique can also be used to establish the sameness of registration batches produced by different manufacturing procedures or at different manufacturing sites.
Post-approval IVRT can be used to ensure quality of production and support site or other changes to a product. The method optimises formulation development and contributes time and cost efficiencies by providing predictive estimates in respect to the in vitro performance of a drug, while also ensuring regulatory agencies can be provided with the information they need. Another significant advantage of in vitro studies is the ability to control the conditions of the experiment in ways that are not possible using human subjects. This allows for more standardised procedures in the laboratory.
Conclusion
The value of IVRT in understanding how a developed formulation will perform in delivering a drug to or across biological tissue is increasingly being realised. Its use is increasing as both a development tool and a means of setting product specifications. With IVRT likely to soon become a regulatory expectation, the use of a suitable system is critical to enable the generation of a properly validated method that can ensure the strict control of variables and assess manufacturing quality with time.
References
- US Food and Drug Administration, Guidance for Industry: Nonsterile Semisolid Dosage Forms: Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Release Testing and In Vivo Bioequivalence Documentation: www.fda.gov/downloads/drugs/guidances/ucm070930.pdf (May 1997).
- A. Olejnik, et al., “Active Compounds Release from Semisolid Dosage Forms,” J. Pharm Sci. 101(11), 4032–4045 (2012).