Western blotting has been used for more than three decades as an essential tool in the detection, quantification and monitoring of proteins and protein purity. The technique first arose in the late 1970s and was based on the concept for Southern (DNA) blots, developed by Edwin Southern in 1975, and which was followed by Northern (RNA) blotting in 1977. George Stark, Harry Towbin and W. Neal Burnette all contributed to the development of this technique, although it was Burnette who named the technique the ‘Western blot’ after Edwin Southern, in addition to acknowledging Burnette’s US West Coast location.
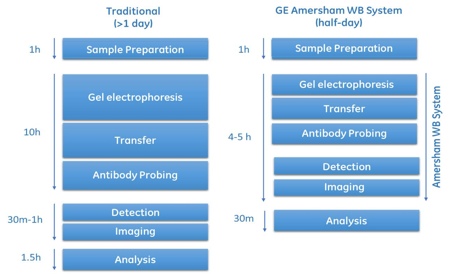
Over the past 30 years, the traditional Western blotting protocol has remained a surprisingly manual technique. A limited number of devices have been available for the automation of individual steps, often with the main focus on speed. Many labs have their own set-up and protocols and some skills are required to obtain high quality results. Western blot workflow involves electrophoresis, transfer, antibody incubation, washing, imaging and analysis of Western blots (Figure 1).






