They are non-specific, prone to low recoveries and very subjective. Furthermore, studies have also shown that some elements give poor recoveries. So, whilst being “easy” to perform, it is clear they are inadequate. Therefore, many analysts have argued that these methods should be replaced by more specific instrumental techniques, thus allowing the specific analysis of individual elements — in a quantitative manner — to enable the sensible scrutiny of the potential toxicity of pharmaceuticals.
After much work and debate, guidelines issued by ICH in 2014 have been formally adopted by the US and European pharmacopoeias; and, from 2018, a harmonised approach to elemental impurity analysis will be in place.
ICH guidance
The guidance issued by ICH lists 24 elements, grouped into four categories based on their relative toxicity, likelihood of occurrence and route of administration. The elements have been assigned exposure limits, given as permissible daily exposure (PDE) limits in µg).
Usefully, the permitted concentration in drug products are also included (as µg, assuming a 10 g daily dose). The ICH guidance has been reproduced verbatim within the European Pharmacopoeia (EP) chapter 5.20 (Metal Catalyst or Metal Reagent Residues), with a compliance date of December 2017 for all products (3 years after the final ICH Q3D guidance was issued). The US Pharmacopeia (USP) has set a final compliance date of January 2018.
ICH and the pharmacopoeias
With general harmonisation obtained, it is now a lot simpler for companies to start their implementation strategy whilst ensuring compliance with the regulations, albeit that the analytical aspect of compliance will probably require finding an accredited partner laboratory capable of doing the testing.
The general approach now accepted for the control of elemental impurities is very much based on a risk assessment. Elements deliberately added to the process are automatically included, as are the four Class 1 and three Class 2A elements (Table I), followed by any of the elements in Class 3 for non-oral dosage forms.
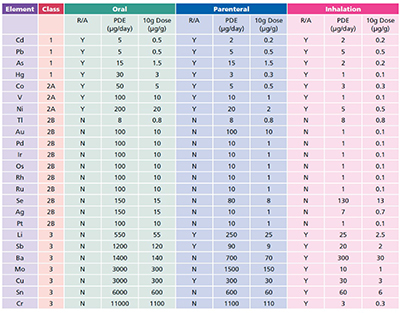
Table I: ICH elements and limits, R/A: Risk Assessment, these are elements that need to be considered as part of the risk element
Risk assessment
As part of the risk assessment, all aspects of the process must be examined, allowing the manufacturer to make an informed decision about the level of testing required to ensure compliance with the regulations.
In most cases, the risk assessment will not return many causes of concern. The impact of manufacturing equipment is generally minimal if the equipment and process is well controlled by standard GMP practices, and water can also be eliminated if the proper pharmacopoeial controls are utilised thoughout the manufacturing process.
Other potential sources to be considered are catalysts, container closure systems and excipients. The latter come from a wide variety of sources and can also be categorised into different levels of risk. For example, a manufactured excipient would be unlikely to be contaminated with metals such as cadmium or mercury, whereas a plant-derived material, such as sucrose, would have a slightly increased risk of having these elements present. Mined excipients are generally of the greatest concern as these materials are often found alongside many other minerals in the ground.
Having identified the risk, it will then be necessary to plan adequate controls; and, in many cases, this will require some analytical testing of the materials, involving the following considerations:

- Raw material or final product testing? Testing a raw material allows for the rejection of batches before they end up in the final product, but the component of risk can be diluted into the final product.
- Will a threshold study be sufficient? Will testing six pilot batches or three full-scale batches at 30% of specification demonstrate minimal risk?
- If a threshold study is not sufficient, or the results are non-conforming, then will the testing be batch release, skip testing or due diligence control with occasional batches tested?
Once the approach has been decided, it will then be necessary to develop an analytical method to do this testing. This process will generally require a feasibility and method development step followed by a validation step.
Method development and validation
One of the key steps of developing a suitable method for elemental impurities testing is the preparation of the samples. The USP <233> chapter usefully sets out three distinct preparation techniques: direct analysis, dissolution and digestion.
Direct analysis is, unfortunately, rarely applicable as aqueous solutions with a minimal matrix are uncommon. The specifics of dissolution and digestion are less important than an understanding that each has its limitations, and both have the potential to cause interferences depending on the elements being tested, and the technique being used. Having three preparation techniques to call on is not the same as saying that all three are equally valid in any given analysis.
Once a suitable digestion technique has been developed, then the samples can be analysed. Typically, this should be done using Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) as it allows for low detection limits to be reached, thus allowing the required specifications to be obtained. These limits may be challenging for alternative techniques, such as Inductively Coupled Plasma-Optical Emission Spectrometry (ICPOES), but ICP-OES may be useful when higher limits are set or a particular interference is encountered when using ICP-MS.
In any event, whichever approach is chosen, the method must be validated to demonstrate that it is fit for purpose. The ICH guidelines and the USP both allow for two different types of validations to take place, namely limit and quantitative. A limit test will only show whether a sample is above or below specification. Quantitative validation allows for a numerical result to be generated, which may be more useful for monitoring and data gathering as part of a risk-based approach to elemental impurity control.
Validation
The decision regarding the extent of the validation will ultimately come down to the needs of the manufacturer and the extent of the testing. A limit test may be sufficient for a threshold study, but a full validation may prove to be more robust if used for batch release testing.
Conclusion
The road to agreement on elemental impurities regulation has been a long and winding one, but it does seem that there is now a clear view to the final destination. The work of the ICH and the relevant local regulators has led to a final procedure that has made the testing requirement less onerous on the manufacturer. With adequate risk assessment procedures, the process of compliance is much simplified and reduces the reliance on analysis.
When the risk assessment does not allow for the elimination of testing, validated methodologies are critical and, by collaborating with an accredited, expert contract laboratory, much of the burden can be reduced.