Vaccines are medicinal products intended to elicit an immune response(s) that can prevent (prophylactic vaccine) and/or lessen the severity (therapeutic vaccine) of a given disease.
Vaccination involves priming the immune system of a host with an infectious agent or components of an infectious agent, modified in a manner to ensure that the vaccine does not cause any harm or disease to the host, but ensures that when the host is confronted with that infectious agent, its immune system can respond adequately to control the invading organism before it causes any ill effect.1
Like other non-vaccine pharmaceuticals, the development of vaccine is also a step-wise process comprising preclinical proof of concept, non-clinical development (efficacy, quality and safety) and clinical development; the data generated in the initial stages guides the strategy for the next stage, until it is proven to be efficacious and safe in the well conducted clinical trials in the target human population.
Special features of vaccines from a safety perspective
The decision to approve a drug is taken after careful assessment of the potential risk that the product may pose to the patients and the benefits that the products provide in terms of preventing or treating a disease. Therefore, a drug product may get approval even if it is known to cause some harm, provided that the relief that the patient gets will far outweigh the risk, taking into consideration other existing medical options.

Sebastian Joseph
With respect to safety evaluation of vaccines, particularly preventive ones, two important things need to be kept in mind:
- first, the safety bar is very high; vaccines are given to healthy people (at least at the time of vaccination) as opposed to patients and are given to millions of people in a short time span
- secondly, some sections of the general public still hesitate to get vaccinated, partly because of the misconceived notion about their potential to cause an adverse effect.
Therefore, the safety evaluation strategy, both non-clinical and clinical, should be robust enough to identify the potential risk and to show sufficient evidence of safety.
How do I plan my non-clinical safety evaluation strategy?
Potential safety concerns associated with vaccines include general systemic toxicity (paradoxical) enhancement of the intended disease, the induction of local toxicity, pyrogenicity, adverse immunological effects such as autoimmunity or sensitisation and, in some cases, teratogenicity/reproductive effects.
There cannot be a single strategy that can meet the requirements of every type of vaccine under development. With the advancement in science, new types of vaccines are being developed, such as DNA, mRNA and those involving recombinant viral vectors and recombinant proteins, etc.
To complicate the matter further, various types of adjuvants, antigen combinations, cytokines, complex excipients, etc. are being used, and different delivery methods are explored with an intent of developing most efficacious and safer vaccines.
Therefore, vaccines represent the most diverse class of product candidates in the pharmaceutical industry. Although the basic principles laid out for the safety evaluation of non-vaccine pharmaceuticals applies to vaccines, there are fundamental differences between vaccine and other pharmaceuticals that necessitate a careful, tailor-made strategy that suits the individual type of vaccine being developed.
A checklist-based study using a standard design is unlikely to satisfy either the scientific or regulatory requirements; rather, the strategy should be designed to account for the type of vaccine being developed, the nature of the antigen, the type and duration of the intended immune response, potential similarity/dissimilarity in the immune response between non-clinical species and human, the route and method of administration, and the qualitative and quantitative composition of the vaccine product, including adjuvants, excipients and potential impurities.
Regulatory expectations and the study requirements
Guidelines with respect to the development of different vaccines are available from agencies such as EMA, US FDA, ICH and WHO (see Table I). These guidelines provide a general framework for evaluation and it is unlikely that all the studies included in the guidelines would be needed for the candidate vaccine.
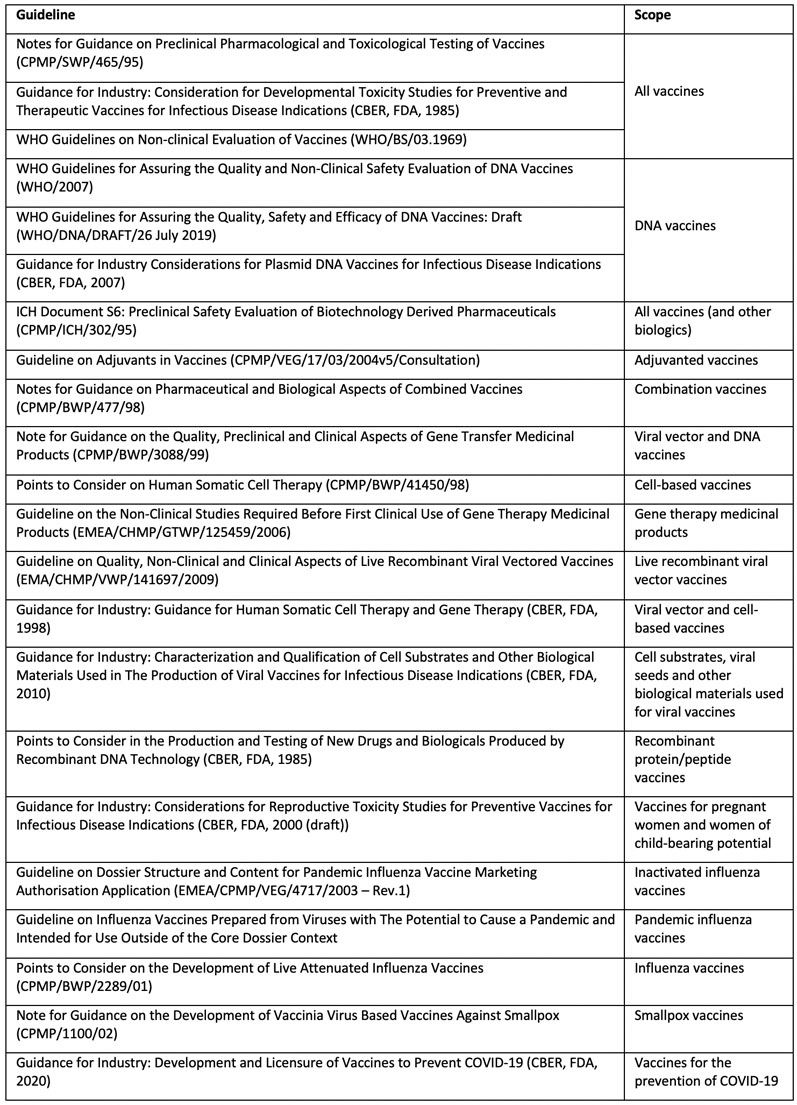
Table I: Guidelines relevant to safety testing of vaccines
It is also true that studies/investigations not listed in these guidelines may also be warranted depending on specific issues related to the vaccine being developed. Regulatory agencies also likely to take a case-by-case and science-based approach when it comes to identifying the studies needed, their design and timing.
Therefore, engaging the concerned regulatory agency in a timely manner — and seeking their feedback and concurrence on the proposed strategy early in the development process — avoids delays in development/approval owing to concerns raised by the agencies that may requires additional investigations.
Fundamental principles and the studies needed for the non-clinical safety evaluation of any pharmaceuticals are laid out in ICH M3.2 Certain concepts described in ICH M3 do apply to vaccines, however, owing to the unique features of vaccines; there are considerable differences with respect to type of studies needed and their design as summarised in Table II.
Pharmacodynamic endpoints are critical to enhance the study value and acceptance
As the vaccine is inherently designed to act on the immune system, the evaluation of effects on immune organs and/or functions are invariably included as part of any toxicological studies. These evaluations are important from a safety assessment perspective.
However, evidence of immune responsiveness as a measure of pharmacodynamic effect in a toxicology study is an important entity that enhances the acceptability and value of the study performed. This is especially important when the tested vaccine has not produced any adverse effects, especially on the immune system.
This evidence of immune responsiveness is determined by antibody level and time course or other markers of immune activation (such as specific T cell type activation). The evidence confirms that the chosen species is relevant to the vaccine under development and therefore the effect profile in the non-clinical studies is likely to be relevant and predictive of response in human.
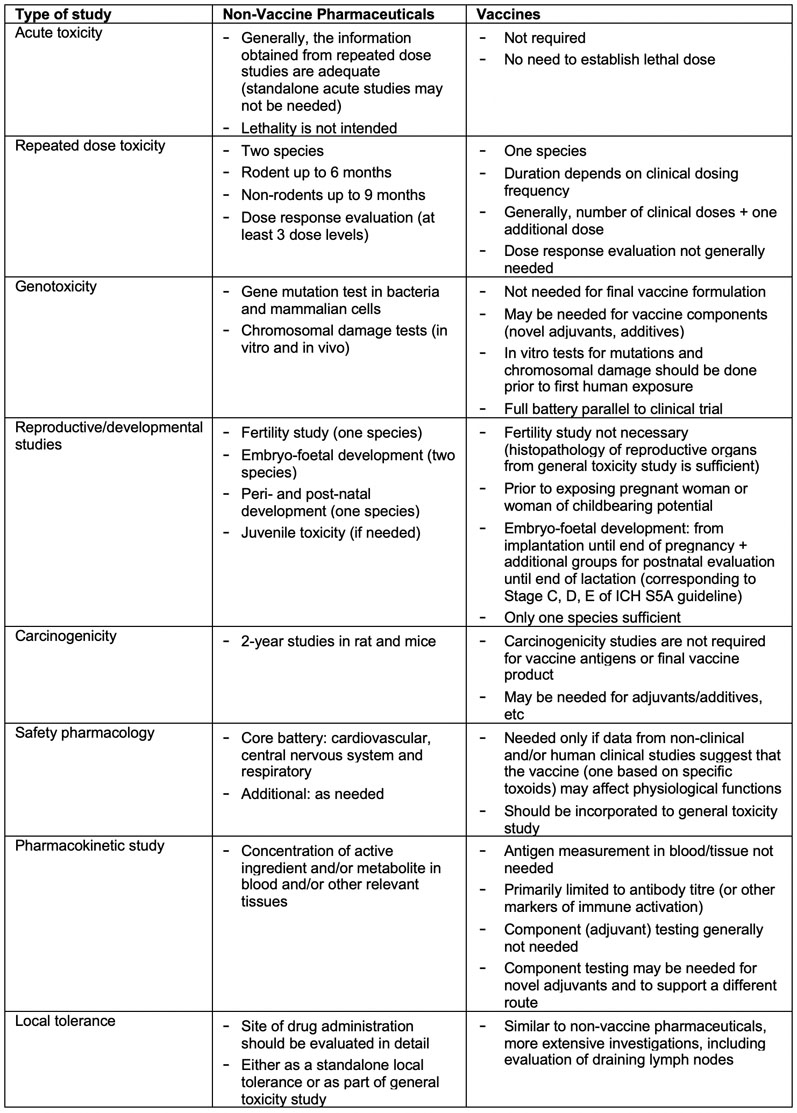
Table II: Non-clinical safety evaluation of candidate vaccine: regulatory requirements vis a vis non-vaccine pharmaceuticals(2–5)
Novel additives require extensive toxicological assessments
The risk of adverse effects with the additives used in a vaccine, including adjuvants, excipients and preservatives, should be evaluated and their level in the vaccine should be justified. Additives previously used in medicinal products, for which safety has been well established, may not require additional testing.
However, the context in which it is used in the candidate vaccine may be different from the previous products. Therefore, a careful evaluation of all available data, either proprietary or publicly available, should be used and a decision taken whether any new studies are needed.
If there are certain data gaps, it is possible to bridge them by including an additional additive-only group in the toxicity studies that are planned for the vaccine. However, standalone studies investigating genotoxicity, teratogenicity, target organ toxicity, sensitisation, etc., may be warranted depending on the vaccine type and the target population.
In such cases, most of the studies recommended in the ICH M3 guideline may be applicable. For adjuvants, in addition to assessing the safety by itself, it is also important to assess whether the combination of antigen and adjuvant exerts a synergistic adverse effect in the animal model … and when species-specific proteins (such as cytokines) are used as novel adjuvants, the issue of species-specific responses should be considered.6
References
- www.who.int/biologicals/areas/vaccines/dna/en.
- www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals.
- www.ema.europa.eu/en/ich-s6-r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals.
- www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-developmental-toxicity-studies-preventive-and-therapeutic-vaccines-infectious-disease.
- www.who.int/biologicals/publications/trs/areas/vaccines/nonclinical_evaluation/en.
- www.who.int/biologicals/areas/vaccines/ADJUVANTS_Post_ECBS_edited_clean_Guidelines_NCE_Adjuvant_Final_17122013_WEB.pdf?ua=1.