Gene therapies enable the delivery of a functional gene to a patient to treat and correct a genetic disease by replacing or compensating for a dysfunctional gene (Figure 1). One of the more common systems for gene delivery uses a non-replicative viral particle, called adeno-associated virus (AAV).
Designed to be just a one-time treatment, gene therapy offers potential solutions to many genetic diseases.
With this promising therapy, though, comes challenges. Companies must overcome a host of analytical challenges in addition to streamlining processes to ensure that this therapy can become a viable option in the future. Here are some key considerations that developers should think about as they move forward with their gene therapy development.
Determining empty versus full capsid ratios
Each AAV particle, referred to as a capsid, should deliver DNA encoding the desired gene. Yet, one major challenge associated with large-scale manufacturing of the AAV vector is the production of impurities, such as empty or partially empty capsids (Figure 2).
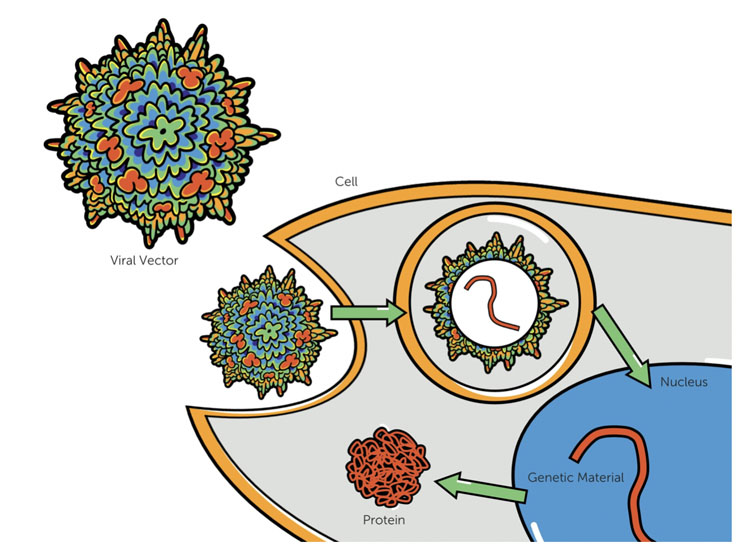
Figure 1: In gene therapy, viral capsids containing the functional gene are delivered to the patient’s cells where the genetic material is then delivered to the nucleus and expressed as a protein. This differs from cell therapy wherein living cells, which may or may not be genetically altered, are delivered to the patient
When empty or inactive capsids are given to a patient, there can be competition for gene delivery with full and functional capsids, which can lead to increased dosing to achieve the desired therapeutic effect. Unnecessarily high dosing brings the increased risk of potentially life-threatening adverse effects, such as immunotoxicity.2
AAV impurities are reduced during downstream processing using analytical ultracentrifugation or chromatography. Analytical methods are used to measure effectiveness of the purification.
Analytical ultracentrifugation is a traditional method; however, high-pressure liquid chromatography (HPLC) is becoming more common with the emergence of BIA Separations’ CIMac AAV full/empty analytical columns. Another added benefit to HPLC is the small sample volume requirement and robustness of the assay.
Detecting additional impurities
It is also crucial that a developer’s analytical partner can test for the large range of impurities that may arise during therapeutic development. In addition to the empty AAV capsids, mentioned above, there are many other product-related impurities that could arise during development, such as bioprocess intermediates, host cell protein contaminants and degraded or aggregated forms of the vector product.2
When performing viral protein purity testing on the vectors, capillary electrophoresis has been shown to be highly effective for CMC purposes. This quantitative method offers a high precision, robust and sensitive way to confirm the expected ratio of viral capsid proteins and detecting unwanted protein contaminants.
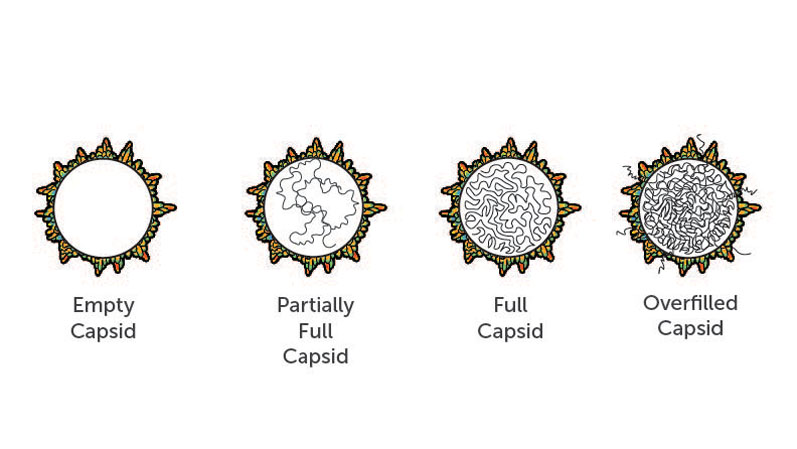
Figure 2: Overfilled capsids, which contain too many copies of the gene, partially full capsids, which contain lower copy numbers or truncations of the gene, or empty capsids, which contain no genetic material, are all problematic and can cause adverse effects in gene therapy patients
An analytical partner will also need to be well versed in detecting and quantifying various other impurities, including
- residual polyethylenimine (PEI): PEI is used during the generation and purification process for AAV; this is best detected with HPLC
- residual iodixanol: used to generate density gradient and purify complex biological materials, such as viral particles, this is best detected with HPLC
- residual benzonase: used to digest the residual DNA or RNA, which often come from lysed host cells used to produce AAVs, this is best detected with ELISA or selective ion monitoring mass spectrometry.
Keeping costs down and efficiency high
Whereas gene therapies promise to drastically improve the lives of those with genetic diseases, they are expensive and complex to develop.3 These therapies are also expensive for patients because they are a one-time treatment, so the cost of a single therapy may be high because it potentially means a lifetime of benefit.
It is important for developers to work with an analytical testing partner that can streamline this process for them to reduce the burden on both developers and patients.
Currently, there is a lack of standardisation that causes difficulty when comparing inter- and intra-lab variability in strength, potency and particle-to-infectivity ratio of AAV-based gene therapies. There are currently two AAV serotype reference standards available.4
Now there is a call to create more robust assays that can ensure quality control consistency to help address this concern.
Guidance from the US FDA is frequently changing because of the newness of this type of therapy. In January 2020, the agency released seven new guidelines for gene therapy manufacturing and clinical development.5
These are seen as a minimum requirement for those working in the field and gene therapy developers are pushing themselves to achieve a higher quality standard than what is required and share their learnings with the industry. These efforts will help to bring gene therapy to a new level of safety.
The future of gene therapy
Gene therapy offers so much potential and the field is moving forward quickly. Improving manufacturing efficiency and sharing learnings across the industry will help to make these treatments more accessible to the patients who need them. As regulatory bodies continue to become more familiar with gene therapy, the road to commercialisation will become more efficient as well.
References
- W. Colasante, et al., “New Approaches To Market Access And Reimbursement For Gene And Cell Therapies,” Cell & Gene (2018): www.cellandgene.com/doc/new-approaches-to-market-access-and-reimbursement-for-gene-and-cell-therapies-0001.
- J.F. Wright, “Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment,” Biomedicines 2, 80–97 (2014): doi:10.3390/biomedicines2010080.
- R. Stein, “At $2.1 Million, New Gene Therapy Is The Most Expensive Drug Ever,” NPR (2019): www.npr.org/sections/health-shots/2019/05/24/725404168/at-2-125-million-new-gene-therapy-is-the-most-expensive-drug-ever.
- ATCC Virus Reference Materials: www.atcc.org/en/Standards/Standards_Programs/ATCC_Virus_Reference_Materials.aspx#.
- US FDA, “FDA Details Policies on Gene Therapies in Seven Guidances,” www.fdanews.com/articles/195767-fda-details-policies-on-gene-therapies-in-seven-guidances (2020).