A cornerstone of any molecule’s long-term success is its early phase development. Having the needs of the finished product in mind from the outset of a project, including how the molecule will be manufactured and ultimately formulated, can help to avoid potentially significant additional costs and delays at a later stage.
There are many reasons why small molecule drugs fail in clinical trials; by contrast, there are some measures that can be taken during early phase development to minimise future setbacks. Six factors that organisations should carefully consider are funding, analytical capabilities, solid state science, regulation, hazard evaluation and a full-lifecycle perspective.
Funding and effective spending
Current estimates typically put the time that a molecule takes in preclinical discovery and development at 3–6 years and the cost of progressing a project from discovery to commercialisation at up to $2.8 billion.1,2
Annual global pharmaceutical R&D spending is now upwards of $186 billion, partly because of the rise in therapeutic complexity and increased costs of biologic and advanced medicines (such as cell and gene therapies).3
One study showed that 22% of failed Phase III candidates resulted from insufficient funding; securing adequate finance for a new project can be challenging, particularly for smaller or relatively unknown biotech companies focusing on a single molecule.4

Early phase development is often about speed of progression and there could be limited data or results to show investors that may be keen to see a potential return on investment. A credible and experienced outsourcing partner can offer guidance and advice to innovators regarding how to be resourceful and prioritise the allocation and most effective use of capital during multiple fundraising rounds.
The early development phase often involves intensive research and analytical work; and, for companies with limited resources or expertise in these areas, outsourcing can offer the best method of meeting these needs.
Satisfying stringent regulatory requirements, characterising the product’s properties and affirming its safety and efficacy require a comprehensive analytical approach and experienced scientists using specialised equipment.
Safety and analysis
Product safety is paramount: 17% of Phase III trials fail because of safety reasons, making this the second largest cause of failure after lack of efficacy.4 Target identification, method development and process validation are key analytical steps that must take place early in any project — as this information will be carried through the entire development process.
Tailoring the analytical strategy to meet the organisation’s specific project requirements and priorities will ensure that all the necessary data are compiled whilst adhering to time and budget constraints.
When developing a process, chemists must be aware of concerns about any raw materials/intermediates that are classed as potential genotoxic impurities (PGIs).
M7 guidelines released by the International Council for Harmonisation (ICH) in 2017 advise on the assessment and control of mutagenic impurities; any early phase synthetic route must ensure that, as a molecule moves through the clinical phases, the risk of patient exposure does not reach dangerous levels.5
This is also true for elemental impurities, meaning that an appropriate strategy for analytical method development is necessary.
The route of synthesis can sometimes be changed to avoid the use of reagents or materials that are themselves PGIs or elemental impurities; but, as this is not always possible, priority should be given to the development of a robust analytical process to ensure any impurities can be detected accurately within specifications.
Solid state properties of the API
The early identification and development of the API’s optimal solid form is another key factor; this has a significant impact on solubility, bioavailability and other properties affecting safety and efficacy. This is especially important if the API is in Class II or IV of the biopharmaceutics classification system (BCS) and has low aqueous solubility (Table I).6
Salt and cocrystal screening may identify a version of the API that has improved solubility and can increase bioavailability. Understanding the polymorph landscape of an API can both identify a preferred form and also assist with development. These investigations will guide the developability of the API as well as drug product formulation.
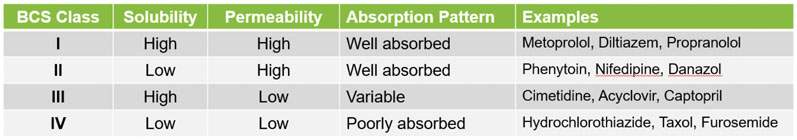
Table I: Solubility and permeability characteristics of APIs
An outsourcing partner with strong solid-state chemistry capabilities and expertise can tailor salt and cocrystal investigations to meet client requirements. This might require a focused salt or cocrystal screen of the API with coforms that may increase the melting point or enhance aqueous solubility, or a broader screening programme of the behaviour of an amorphous API in various conditions.
Approximately 90% of small molecule drugs are commercialised in their crystalline form, but achieving the desired solid state may require a variety of different investigations — including salt, cocrystal and polymorphs — before crystallisation development.7
Solid state screening should be completed (at the latest) before a drug enters Phase III clinical trials and the generation of the registrational stability batches (whichever is earlier). If the initial batches of API are generated before a polymorph screen, the solubility and stability of the drug substance could be affected and may need to be re-established.
Although it can be a time-consuming process, establishing the ideal solid form of an API early in the lifecycle avoids potential challenges with the formulated drug product. Having to change the solid form profile during clinical development means having to repeat many of the previous steps, running the risk of negatively impacting both costs and timelines.
Regulatory assurance
Before a product enters clinical trials, an investigational new drug (IND) application must be submitted to the US FDA that affirms the product’s safety and efficacy by supplying all the necessary supporting data and manufacturing information.
Again, addressing regulatory requirements in the early phases of a project, and not having to make changes later on during development, could avoid setbacks and additional costs.
An effective Chemistry, Manufacturing and Controls (CMC) strategy will ensure ongoing quality and compliance as the project scales-up, all while striking a balance between speed and cost.
To achieve this, certain process optimisation steps are often reserved until after the preclinical stage; here, working with an experienced partner can help to ensure that resources are allocated efficiently as the product progresses through trials, maximising its chance of success while managing budgets and timeframes.
During early phase clinical development, specifications for raw materials, intermediates and the API will need to be defined, as will the appropriate testing methods. As the programme progresses, test methods/details will be added and tolerances may be tightened as process knowledge is gained.
Raw materials will need further testing as opposed to relying on suppliers’ Certificates of Analysis for quality release in early phase … and analytical validation will be required.
During this evolutionary period, test methods/specifications of the drug substance will continue to be updated in Module 3 of the common technical document (CTD) in preparation for the final new drug application (NDA) submission.

Safety and hazard evaluation
Although an API can be synthesised safely in the laboratory, using certain reagents or solvents may give rise to challenges as the scale of synthesis increases.
Pitfalls that could present setbacks at a later stage can be avoided by working with a partner that has chemistry process development experience, is able to implement measures to eliminate or substitute materials and processes that may be unsuitable for scale-up and eventual commercialisation, and that can continually monitor potential hazards throughout the project lifecycle.
The earlier changes are made in the process, the less need there may be to duplicate work in terms of the potential impurity specification, analytical testing needs and toxicological profile of the API.
Long-term vision: a full lifecycle approach
The early phase considerations outlined above should inform every decision in the API’s lifecycle to enable the proactive mitigation of challenges that could surface as the project moves into later stages and, ultimately, on to commercial approval.
An experienced outsourcing partner with the flexibility, expertise and facilities to take projects from the laboratory to commercial scale can maintain critical project continuity and minimise time-consuming external tech transfers.
Furthermore, a partner with the infrastructure in place to handle special considerations, such as controlled substances, can eliminate the need to transfer a process to another provider, avoiding additional delays.
There is no “one-size-fits-all” approach to drug development, as every molecule is unique. Early phases require a difficult trade-off between generating results and data, and balancing the time and cost needed.
Ensuring the efficient use of resources can be assisted by using outsourcing partners with the ability to fully understand project requirements, reputation, cost-effectiveness and risk evaluation.
By prioritising timeliness and quality against short-term efficiencies and long-term requirements, the additional experience that a development partner can bring should also increase the quality of drug candidates being progressed towards the clinical phases and reduce the risks of failure.
References
- www.sciencedirect.com/science/article/abs/pii/S0167629616000291?via%3Dihub.
- http://phrma-docs.phrma.org/sites/default/files/pdf/rd_brochure_022307.pdf.
- www.statista.com/statistics/309466/global-r-and-d-expenditure-for-pharmaceuticals/.
- www.ncbi.nlm.nih.gov/pmc/articles/PMC6092479/.
- www.ema.europa.eu/en/ich-m7-assessment-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential.
- https://doi.org/10.1023/A:1016212804288.
- https://aiche.onlinelibrary.wiley.com/doi/full/10.1002/aic.11555.