Microbial biotechnology, in parallel with electronics, computing and data analysis capabilities, has been through a step change in capacity during the last 25 years. This has resulted in the ability to perform tasks in a few weeks or months that would have previously taken a team of scientists and technicians a considerably longer time to complete.
In this case study, David Gervais, Head of Product and Fermentation Development at Porton Biopharma Limited (PBL) describes how a non-GMP enzyme manufacturing process has been successfully converted from native production to recombinant using bioinformatics and next-generation sequencing (NGS) to facilitate the change.
The challenge was the conversion of the production of an enzyme product from a native Pseudomonas system to a recombinant system in Escherichia coli. For this product, the development and alteration of the manufacturing process was done in a quick and efficient manner, leading to vast improvements in yield and method of production.
At the outset of the project, the sequence of the native enzyme, which has been produced for decades using a non-characterised bacterial strain, was not known. This is not uncommon for enzyme products developed in the 1970s and 1980s prior to the introduction of recombinant technology.
In addition, such production methods would now be considered to be quite antiquated and can suffer from low expression yields in native organisms. To resolve this, whole-genome sequencing and in silico gene identification techniques were used, followed by total synthesis of the gene sequence.
The resulting gene was successfully transformed into E. coli and the target enzyme expressed, purified, and verified in terms of sequence and performance in the intended application. The entire process, from initiation to proof-of-concept data, took only a matter of months, and demonstrates that modern bioinformatics can be used to drive innovation in process development for recombinant proteins and enzymes.
Recombinant expression system
At the start of the project, the gene encoding the Pseudomonas enzyme of interest was not known. The enzyme is derived from a novel strain of Pseudomonas sp. held within the stock of Porton Biopharma at Porton Down. For the rapid turnaround of this project, a quick method of gene identification was required that was more in line with next-generation sequencing (NGS) techniques than traditional polymerase chain reaction (PCR) methods.
Therefore, shotgun sequencing was used on the whole organism genome, along with identity modelling and gene fishing in silico. For the identity modelling and gene fishing, the resulting shotgun sequence results were aligned with published sequences of similar enzymes from closely related organisms that were likely to match the unknown gene query.
A likely sequence of the correct gene length was identified that had 87% identity to closely related enzymes from the literature. This gene sequence was sent to a third-party gene synthesis provider and cloned into several isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible expression vectors with either C-terminal or N-terminal hexahistidine purification tags.
The resulting plasmids were transformed into competent E. coli cells and grown on selective media. Cells were grown in Terrific Broth (TB) containing kanamycin for selection, and expression of the target gene was induced by the addition of IPTG. Cell stocks were also created for storage at –80 °C using glycerol as a cryoprotectant.
Initial studies in shake flasks with these cell stocks demonstrated high expression of the target gene. Lysates from these studies were analysed using sodium docecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and a significant band was observed at the target molecular weight.
This band was excised and analysed using N-terminal sequencing (first 20 residues) and found to have 100% identity to the target protein sequence, giving confirmation that the expression system was working and the enzyme was being produced. The target genes stored in frozen cell stocks were also checked by sequencing to doubly ensure that the identity of the target enzyme was intact.
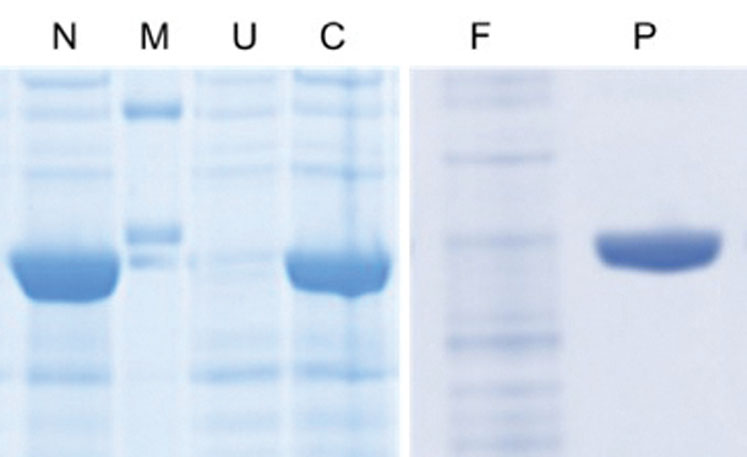
Figure 1: SDS-PAGE analysis of recombinant enzyme production (N = N-terminal His6 expression culture, M = molecular weight marker, U = uninduced culture, C = C-terminal His6 expression culture, F = purification flow through, P = purified enzyme)
The yield of the enzyme in expression cultures was established using the existing analytical method for enzyme activity. Although both versions (N and C-terminal hexahistidine tags) of the enzyme expressed well in E. coli (Figure 1), it was found that the N-terminal version had a 60% higher activity yield, suggesting that the C-terminal version was producing a misfolded enzyme. For this reason, the N-terminal construct was taken forward for process development studies.
Production process development
Having established the recombinant expression system and created frozen cell stocks, an enzyme production process was quickly developed. This involved initial screening of the recombinant construct in shake flasks, followed by 1 L fermentation optimisation runs using a modern system with disposable bioreactors.
This approach allowed the optimum conditions of the fermentation to be quickly screened, so that expression of the recombinant product was maximised. Conditions that were determined included the fermentation temperature profile, agitation speed, induction time, total fermentation time and induction IPTG concentration.
Once a prototype fermentation process was developed at 1 L scale, the process was scaled-up using stainless steel fermentation vessels at working volumes of 10 and 15 L. With this strategy, it was possible to calculate the likely final scale required for routine production and enable a good comparison of the recombinant production system with the earlier, native system. Excellent yields were achieved, as discussed below.
A purification strategy for the enzyme was also developed. The earlier native production process involved a series of traditional purification steps, including cell lysis and centrifugation followed by ion-exchange chromatography (IEX) and tangential-flow filtration (TFF). This process resulted in a less-than pure product that had sufficient enzyme activity for the intended application … but we were confident that this could be significantly improved.
The redeveloped purification process has taken advantage of the purification tag to enable a quick process that delivers increased yields of high purity enzyme. Cells are lysed by sonication or high-pressure homogenisation and subsequently bound to an immobilised metal affinity chromatography column packed with a Sepharose 6 Fast Flow charged with Ni2+ cations. The hexahistidine purification tag, covalently attached to the N- or C-terminus of the target enzyme, binds to the immobilised metal cation and adsorbs the enzyme to the column.
The column may be selectively eluted with imidazole and product fractions collected, affording high purity as depicted in Figure 1. An important consideration is the intended end use of a product and whereas such a method may not be suitable for pharmaceutical production, owing to the potential presence of trace levels of toxic nickel in the product, it is ideal for non-GMP, non-pharmaceutical (and non-veterinary) products such as this enzyme.
Comparison of native and recombinant production
A comparison of the recombinant production system with the native one (Table I) demonstrated the enhanced performance of the former. Unsurprisingly, in addition to the improved yield, the recombinant system had a superior enzyme output at a smaller overall scale, reducing the production complexity and validating the approach taken.

Identification of the target gene, including sequencing and in silico analysis, took approximately one month. After the gene was identified, converting the process from native to recombinant was achieved in approximately three months.
Some further process optimisation was done once the proof-of-concept was demonstrated. Functionally, the recombinant enzyme has now been demonstrated to be equivalent to the native material in terms of performance in the intended application.
Delivering an improved process
The output of such a rationally designed approach is that a key non-GMP enzyme product was successfully converted from native production to a recombinant system in a few months. The gene sequence of the target enzyme was not known, so whole-genome sequencing and in silico gene identification was used, along with synthesis of the target gene. The synthetic gene was transformed into E. coli and used for expression and purification studies.
The recombinant enzyme performs identically to the native enzyme in the target application and affords greater yields compared with the native system. In conclusion, the application of modern molecular biotechnological and purification techniques, in parallel with a rational process development protocol, has been demonstrated to deliver significant improvements in yield, purity and activity. This principle is applicable to other legacy products across the industry.
Bibliography
- D. Gervais, et al., “Validation of a 30-Year-Old Process for the Manufacture of L-Asparaginase from Erwinia chrysanthemi,” Bioprocess and Biosystems Engineering 36(4), 453–460 (2013).
- S. Charlton, et al., “A Study of the Physiology of Bacillus anthracis Sterne During Manufacture of the UK Acellular Anthrax Vaccine,” Journal of Applied Microbiology 103(5), 1453–1460 (2007).