
Acting at the interface between genes and proteins, therapeutic oligonucleotides are a class of drug based on the structure of DNA and RNA. By selectively binding genetic material such as RNA, they influence the expression of targeted proteins to elicit a beneficial therapeutic effect.
Oligonucleotides based on natural building blocks, whether based on DNA or RNA, are rapidly broken down inside cells by enzymes.1 This has driven the need for chemical innovation to modify and stabilise these structures, which are typically oligomers comprising 20–30 nucleotide residues.
Some of these products are single stranded whereas others are double stranded. The synthesis of such molecules, combining natural and unnatural residues, has therefore presented many challenges during the discovery and development process, particularly when scaling-up oligonucleotide synthesis.
In addition to metabolic stability, the delivery of large and unwieldy oligonucleotides to their required site of action is also challenging, necessitating further chemical modifications and tagging strategies.2 This article discusses how enzymatic approaches can contribute to better and cheaper methods for oligonucleotide synthesis.
The chemistry of oligonucleotide synthesis
DNA or RNA consists of deoxyribose or ribose sugar units — with their characteristic bases (A, C, G, T or U) — linked together by phosphodiester bonds.
Traditional oligonucleotide synthesis uses phosphoramidite chemistry and comprises a repeating sequence of four reactions (deprotection, coupling, capping and oxidation) with single nucleotides added sequentially (Figure 1).
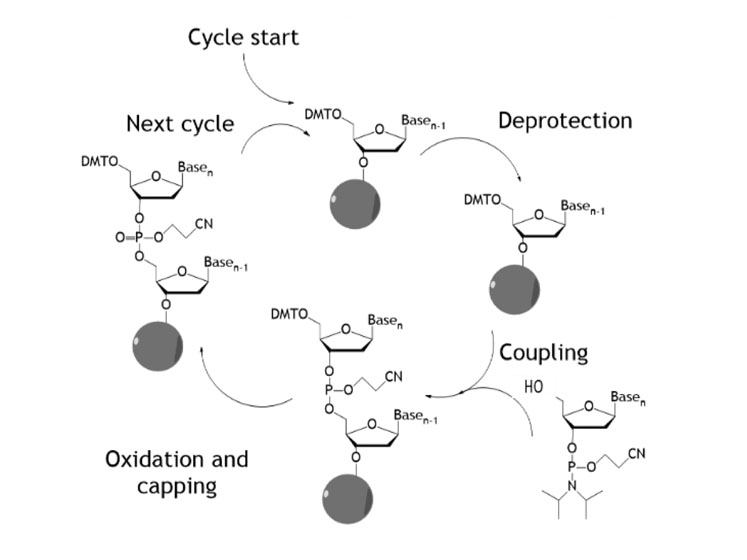
Figure 1: Traditional oligonucleotide synthesis
This is often performed in a solid phase mode wherein the oligonucleotide is extended via this cycle from a solid support. This chemistry has been adapted in numerous ways to allow unnatural monomer units to be incorporated into the oligonucleotide chain.
Various chemical approaches have been adopted to improve both stability and biological performance … and three exemplars (although there are many more) are shown in Figure 2 for derivatives of RNA.
One of these approaches uses a modified phosphodiester backbone during which sulphur is introduced to replace oxygen in the phosphodiester bonds.
A second approach introduces different chemical functionality to the ribose ring, usually in the 2’ position, with methoxy, methoxyethyl and fluoro being the groups most frequently used. A third approach “locks” the sugar ring conformation through the formation of bicyclic structures.
All of these can slow down the rate of metabolic breakdown of oligonucleotides to an acceptable level for effective therapeutic action.
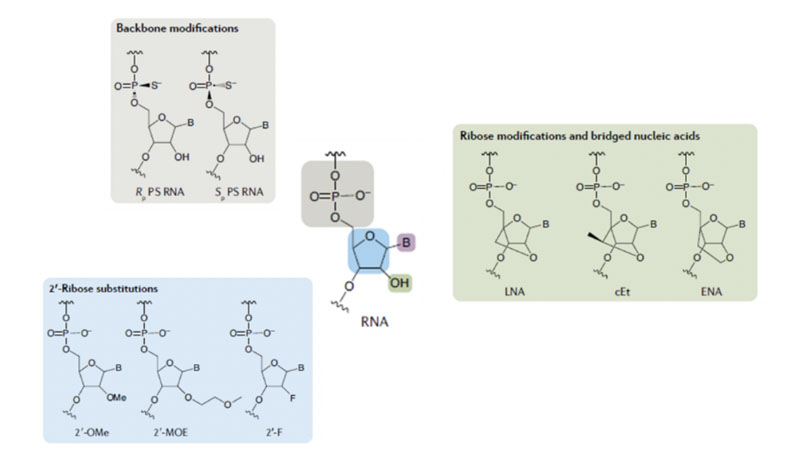
Figure 2: Examples of modification to facilitate oligo stability
As an oligonucleotide approaches market launch, there is a requirement to scale-up and synthesise kilo to tonne quantities of these building blocks and link them together in the correct sequence.
However, large-scale oligonucleotide chemistry can be problematic. For example, the requirement for complex chromatography and its automated hardware can add significant cost to this active ingredient of the drug product.
Perhaps worse, the chemistry is linear and sequential, resulting in a geometric decrease in yield as the chain gets longer.
For example, if the yield for each nucleotide-linking phosphoramidite cycle was 95%, which would be very respectable for a four-step cycle, then the overall yield for a 20-mer product is only 36%. Impurity formation and removal is also often a confounding issue for the process. There is a need, therefore, for new approaches to this chemistry.
Biocatalytic synthesis is around the corner
Pharmaceutical synthesis has been transformed by biocatalysis in recent years. When enzymes have been introduced, routes have been shortened, costs lowered and processes generally “greened up” with spectacular results in many cases.
Almac Sciences has been at the forefront of this revolution in sustainable synthesis, tapping successfully into the transformative changes occurring in molecular biology and resulting in low-cost enzymes that can be confidently engineered for maximum performance.3–5
A strong oligonucleotide synthesis technology base is now available that includes the ability to produce panels of enzymes for screening, a deep understanding of biotransformation development, process optimisation and biocatalyst engineering at the molecular level.
From these foundations, biocatalysis is now starting to make in-roads into oligonucleotide synthesis with developments and concepts highlighted herein.
Addition of single nucleotides to form blockmers
Blockmers are loosely defined as very short single-stranded oligonucleotides of 4–8 residues that form the basis for the enzymatic synthesis of larger oligonucleotides. In the first example illustrated here, RNA ligase was used to form a series of RNA-based blockmers of four residues by adding a single nucleotide to a trimer (designated ABC).
Almac has applied state-of-the-art synthetic biology design to this process by creating panels of diverse RNA selectAZyme ligases for reaction screening.
The reactions in Figure 3 were achieved by screening the selectAZyme RNA ligase panel for residue X coupling (phosphate protected at the 3’ and 5’ positins), with some enzymes showing good activity for a given substrate. As will become apparent, access to enzyme screening panels is critical to identify good activity and is a recurring theme.
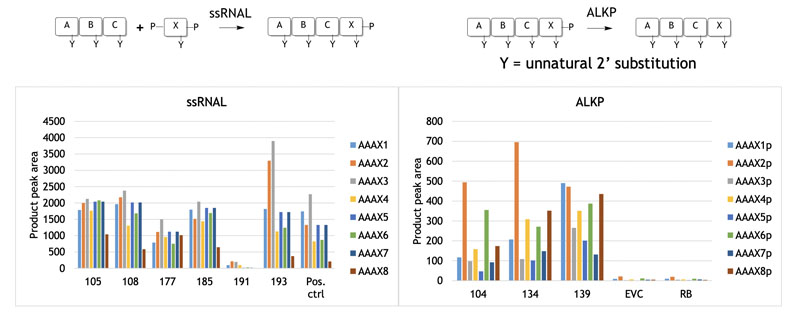
Figure 3: Nucleoside addition to blockmers using selectAZyme ligases
Figure 3 also shows the use of selectAZyme alkaline phosphatase, an enzyme that cleaves the phosphate group off the reaction product at the 3’ position, readying it for a further round of synthesis and chain extension.
This too showed that screening a panel of selectAZyme phosphatases allowed good activity to be identified for the substrates. Once the required blockmers have been assembled, larger strand construction can begin.
Building single stranded RNA oligonucleotides from blockmers
The use of enzymes to construct single strands of RNA by joining blockmers of RNA together was first described in the 1970s.6,7
This approach, which does not rely on a template to guide the addition of incoming substrates, is catalysed by the RNA ligase family of enzymes (as above) and linking together fragments based on combinations of natural and unnatural ribose-based nucleotides. This concept is shown in Figure 4, for example, whereby two unnatural fragments (blockmers) were joined together.
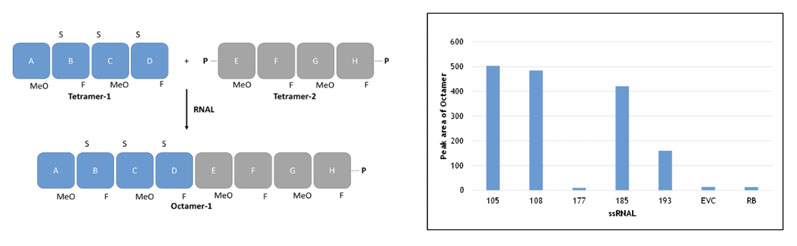
Figure 4: Blockmer to blockmer assembly (non-templated method) using selectAZyme ligases
In this example, the ribose residues both contained a substitution at the 2’ position (unnatural equivalent groups) and were linked together by phosphorothioate esters; the two strategies combined to stabilise the oligonucleotides against degradation.
To develop this successfully, it was critical to have access to a panel of RNA ligases for screening; some enzymes were not active whereas others showed good activity, as can be seen in the insert in Figure 4 (EVC is the empty vector control; RB is reaction buffer only).
Enzyme engineering for ligases
Almac’s approach to enzyme engineering involves using its in-house INSIGHT platform, which allows rational enzyme design from metagenomes at a fraction of the cost associated with traditional directed evolution.
This was applied to RNA ligase to improve the addition of an unnatural nucleotide phosphate — protected at the 3’and 5’ positions (pX2Yp) — to the trimer ABC in the reaction shown in Figure 5.
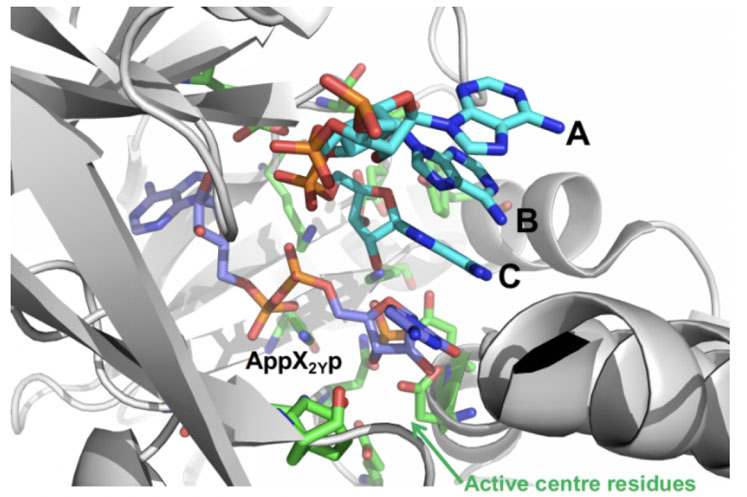
Figure 5: Ligase mediated coupling of ABC + pX2Yp involving mutation of the enzyme RNAL 105
INSIGHT design was used to create a small-but-smart panel of engineered RNA ligases based on an Almac selectAZyme enzyme, RNAL 105, which was redesigned around its active site to allow better interaction with pX2Yp and improved ATP binding; this was needed to drive the reaction.
When the resulting enzyme panel was screened, a near 10-fold increase in activity was achieved compared with the initial enzyme, showing the utility of INSIGHT technology in RNA ligase reactions.
References
- F. Czauderna, et al., Nucleic Acids Res. 31, 2705–2716 (2003).
- I.J. Huggins, et al., Molecules, 24(18), 3287–3300 (2019).
- A.T.P. Carvalho, et al., Phys. Chem. Chem. Phys. 20, 2558–2570 (2018).
- A.T.P. Carvalho, et al., Phys. Chem. Chem. Phys. 19, 26851–26861 (2017).
- D.F.A.R. Dourado, et al., ACS Catalysis 6, 7749–7759 (2016).
- Y. Kikuchi and K. Sakaguchi, Nucleic Acids Res. 5, 591–598 (1978).
- O.C. Uhlenbeck and V. Cameron, Nucleic Acids Res. 4, 85–98 (1977).
For more information
Jill Caswell, Darren Gray, Daniel F.A.R. Dourado and Steve Taylor, Arran Chemical Company, and Tom Moody, Almac Sciences.






