Advanced therapy manufacturers have taken the head start that Nature’s given them and used developments in genetic engineering, computational biology and bioinformatics to perfect the viral particle as a vector for therapeutic DNA and hasten the clinical success of groundbreaking new treatments.
Here, we look into three aspects of viral biology that make them efficient delivery vehicles for gene therapies — and some of the ways that biologists have taken advantage of these properties to improve clinical performance.
When efficient viral infection is a good thing
In normal physiological conditions, maintaining open channels of communication between cells is essential for nutrient transport, signal transduction and other forms of cell-cell interaction.
Viruses are adept at exploiting these channels to hitchhike into cells. For example, the adeno-associated virus (AAV) that’s often used as a vector for gene replacement therapies enters the host cell by interacting with cell surface receptors, leading to endocytosis from clathrin-coated pits.
From there, a change in pH triggers the release of the virus from the endosome, where it’s rapidly trafficked to the cell nucleus.1
Which cells the virus infects and how efficiently it enters them is defined by the specific cell surface receptors it interacts with. This is known as viral tropism.
So far, nine different AAV serotypes have been discovered in human cells and a further four in non-human primates, each of which have different tropisms.2
The first AAV to be discovered was AAV2, which binds to the near ubiquitously expressed heparin sulphate proteoglycan (HSPG), resulting in a broad tissue tropism.3
Other AAV serotypes have different cell surface receptors and therefore different tropisms, which in turn informs the choice of viral vector that’s most appropriate for each gene therapy.
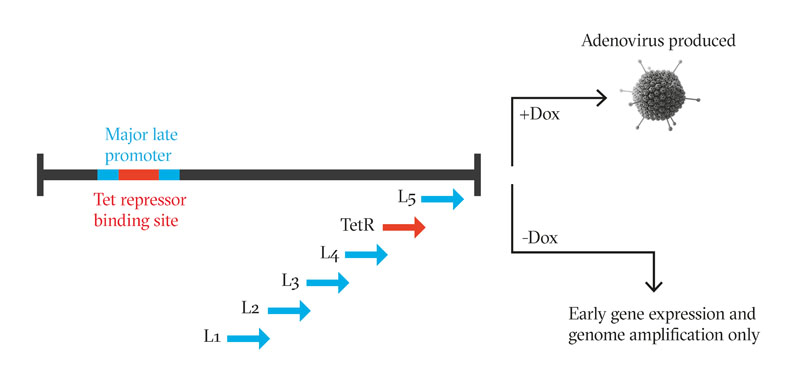
Figure 1: Inserting a Tet repressor binding site into the MLP of the adenoviral genome with a TetR gene downstream introduces a negative feedback loop, in which the adenoviral genome cannot transcribe any late-phase genes that encode the structural proteins in the absence of doxycycline
Despite the divergent tropisms offered by different AAV serotypes, the efficient delivery of gene therapy vectors remains a challenge. Natural exposure to AAV means that large sections of the population have developed neutralising antibodies against certain viral serotypes.
Delivery to the target tissue can be difficult; for example, systemic administration of a viral vector often results in trafficking to the liver, which may not be the intended target.
Access to the target tissue may present another challenge; the virus may get “stuck” on HSPGs and other proteoglycans on the extracellular matrix outside the target cells.4 Indeed, any given AAV capsid may have both advantages and disadvantages for a particular clinical use.
But whatever challenges nature has set for the use of viral vectors to deliver gene therapies, science is striving to overcome them — in this case by capsid engineering to improve transduction in the target cell type, improve specificity of transduction and/or to evade a host immune response.
The first approach to this, termed rational design, relies on understanding the mechanism of transduction and identifying the areas of the viral genome that are responsible for encoding the capsid’s interaction with the cell surface receptor.5
This allows the insertion of the viral genome sequence conferring the desired tropism into the genome of another AAV serotype, which may have other clinical advantages.
This approach has already proved viable in a clinical trial for Duchenne Muscular Dystrophy, in which the capsid region responsible for the efficient transduction of skeletal muscle by AAV1 was engineered into AAV2 (whose safety profile was better characterised and could be easily purified).6

However, the exact mechanism of viral transduction is not yet clear for all AAV serotypes, so a more random approach to capsid engineering, termed directed evolution, may sometimes be necessary.
This involves using the wildtype cap genes as a starting point to create large genetic libraries — either by error-prone PCR to introduce random point mutations into the cap gene or random insertion of peptide-encoding sequences (such as those encoding a known cell surface receptor binder).
Alternatively, mutations can be targeted to hypervariable regions of the capsid, such as surface-exposed loops, which can then be replaced by bioinformatically designed peptide sequences.4 The resulting viral particles can then be selected for transduction of the desired cell type in vitro or in vivo.
When turning the dial on viral genome expression matters
Once they’ve entered the cell, viruses are adept at wresting control of the cell’s DNA replication machinery to force the transcription of their own genome. When the genes that would enable viral replication are replaced inside the virus particle by a therapeutic transgene, it is this DNA that the cell is forced to transcribe.
Almost all gene expression is driven by a promoter: a core region of DNA that’s approximately 100 base pairs upstream of the regulated gene plus an upstream activating sequence or enhancer. Both core promoter regions and enhancers contain clusters of transcription factor binding sites, and transcription factor binding in these areas determines the level of gene expression.
Generally, either promoters that are endogenous to the target cell/tissue type or viral promoters are used to drive transgene expression. Tissue-specific endogenous promoters have the advantage of limiting gene expression to the target cells; however, typically low promoter activity or large promoter size may lead to suboptimal expression levels.
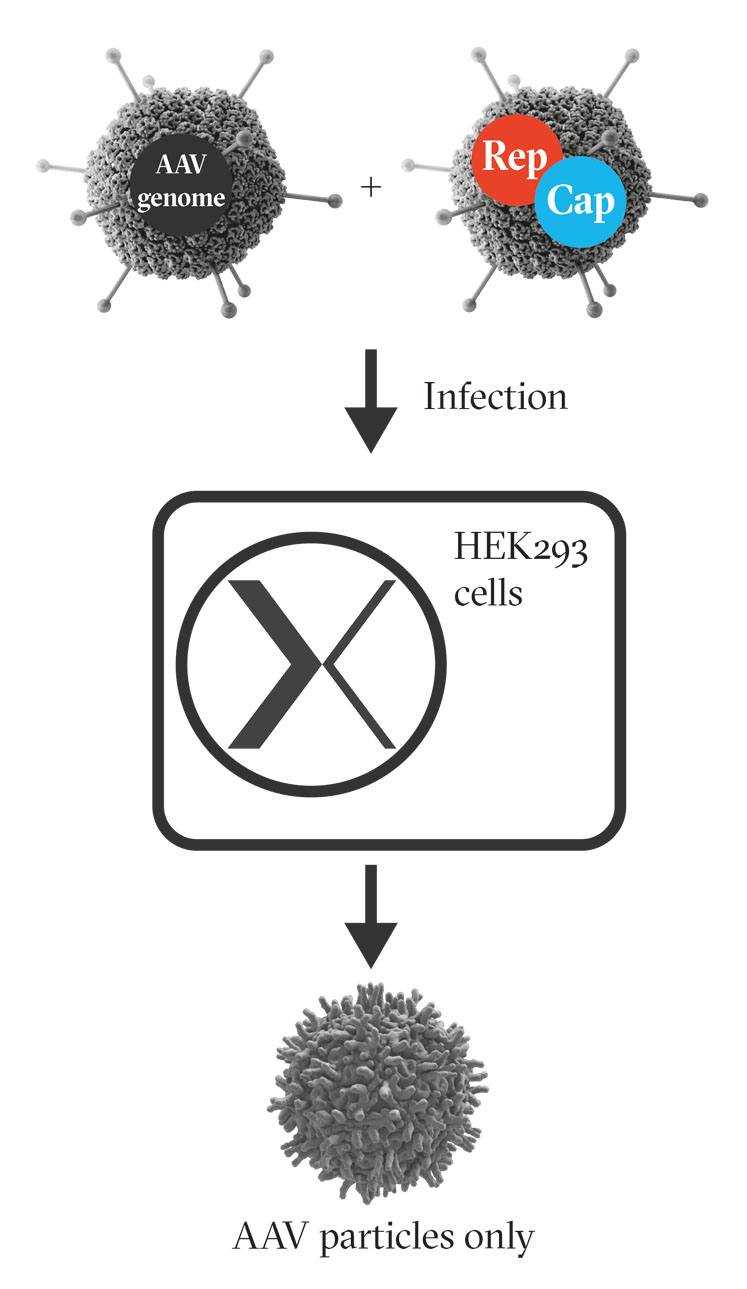
Figure 2: Infecting HEK293 cells with engineered adenoviruses encoding the AAV genome, and AAV rep and cap genes respectively, results in the production of AAV
Viral promoters, by contrast, are constitutively active, resulting in high levels of therapeutic transgene expression in all transduced cells. This carries a risk of toxicity owing to transgene overexpression or stimulation of an immune response due to transgene expression within an antigen presenting cell.7
Engineering the promoter driving transgene expression therefore represents another opportunity to improve the clinical performance of the viral vector.
Error-prone PCR can again be used to insert random point mutations into promoter sequences to change transcription factor binding sites and alter promoter strength. However, this approach is most likely to prevent transcription factor binding and therefore reduce the strength of the promoter.8
Although this may reduce the risks associated with transgene overexpression from a viral promoter, it won’t stop non-specific gene expression. Instead, hybrid promoters built by screening libraries of different promoter and enhancer combinations are likely to present a better option to generate tissue-specific promoters with increased transcriptional strength.8
When harnessing the power of viral genome replication can aid production
Gene therapy manufacture relies on being able to produce huge quantities of viral vectors using cells as mini virus-production factories. However, one of the properties of AAVs that makes them ideal as a vector for gene therapies — its inability to replicate without the help of a second virus — adds challenges for its manufacture.
In nature, this help is provided by adenovirus … but this isn’t a system that’s easily co-opted for clinical production because the adenoviruses must be purified out from the AAV before they’re suitable for clinical use (a complex and expensive process).
To date, the adenoviral help necessary to produce AAVs destined for clinical use is provided by a plasmid encoding an adenoviral genome that’s been pared back to only those genes essential for AAV replication. However, this is a far from ideal process and the complexities and costs associated with plasmid-based AAV production at scale have been widely reported.
Adenoviruses, by contrast, are extremely efficient at replicating themselves once they’ve gained control of the host cell systems. Indeed, following adenovirus infection, almost 90% of the host cells’ mRNA is derived from the adenoviral genome.
Here, viral vector engineering offers an opportunity to harness the power of adenoviral protein production and use it to improve the manufacture of cell and gene therapies.
In this case, scientists have done this with a clever two-step manipulation of the adenoviral genome. The first step was to manipulate the genome in such a way that it could still provide help for AAV production but no longer transcribe adenoviral structural proteins.
This worked because the adenoviral genome is divided into two temporal phases with distinct purposes. All the genes responsible that help AAV to replicate are transcribed in the early phase, whereas the adenoviral structural proteins are all transcribed from late-phase genes under the control of the major late promoter (MLP).
Inserting a Tet repressor binding site into the MLP, and the Tet repressor gene itself into the late region of the genome, essentially cripples adenoviral production; the more the adenovirus tries to transcribe its structural proteins, the more Tet repressor it produces to inhibit MLP activity (Figure 1).
The second manipulation involves inserting either the AAV rep and cap, or the ITR flanked therapeutic transgene, into the early phase of the adenoviral genome. Now, the adenoviral genome can no longer produce adenovirus particles, but infecting cells with two of these edited adenoviral vectors is sufficient to produce high quantities of fully functional AAV (Figure 2).9
Wildtype adenovirus and AAV have coevolved and fine-tuned the supported replication of AAV, so it’s perhaps unsurprising that plasmid-based AAV production cannot replicate the AAV yield or quality of wild-type adenovirus.10
However, this adenoviral genome engineering approach retains all the natural adenoviral helper functions for AAV production, suggesting a manufacturing system that would produce higher yields of more infectious AAV than can be produced by triple transfection.
During the last few years, our understanding of viral biology and genetics has increased exponentially, as has the availability of the tools required to manipulate them.
This is leading to the creation of ever more powerful viral vectors that can be targeted to specific cell types, fine tune the expression of the therapeutic transgene within them and — importantly for the accessibility of these new biologics — be manufactured in greater quantities, at better quality, cost-effectively and with greater ease.
Indeed, the US FDA expects to be approving 10–20 new cell and gene therapies annually by 2025, suggesting that we are entering a new golden era for genetic medicine.
References
- J.S. Bartlett, R. Wilcher and R.J. Samulski, “Infectious Entry Pathway of Adeno-Associated Virus and Adeno-Associated Virus Vectors,” J. Virol. 74(6), 2777–2785 (2000).
- A. Srivastava, “In Vivo Tissue-Tropism of Adeno-Associated Viral Vectors,” Curr. Opin. Virol. 21, 75–80 (2016).
- C. Summerford and R.J. Samulski, “Membrane-Associated Heparan Sulfate Proteoglycan is a Receptor for Adeno-Associated Virus Type 2 Virions,” J. Virol. 72(2), 1438–1445 (1998).
- M.A. Kotterman and D.V. Schaffer, “Engineering Adeno-Associated Viruses for Clinical Gene Therapy,” Nat. Rev. Genet. 15(7), 445–451 (2014).
- C. Li and R.J. Samulski, “Engineering Adeno-Associated Virus Vectors for Gene Therapy,” Nat. Rev. Genet. 21, 255–272 (2020).
- D.E. Bowles, et al., “Phase 1 Gene Therapy for Duchenne Muscular Dystrophy Using a Translational Optimized AAV Vector,” Mol. Ther. 20(2), 443–455 (2012).
- C. Domenger and D. Grimm, “Next-Generation AAV Vectors: Do Not Judge a Virus (Only) By Its Cover,” Hum. Mol. Genet. 28(R1), R3–R14 (2019).
- J. Blazek and H.S. Alper, “Promoter Engineering: Recent Advances in Controlling Transcription at the Most Fundamental Level,” Biotechnol. J. 8(1), 46–58 (2013).
- W. Su, et al., “A Complete “Self-Repressing” Adenovirus System Enables Efficient Manufacture of Adeno-Associated Viral Vectors without Contamination by Adenovirus or Small Drugs,” Molecular Therapy 28, 240–240 (2020).
- N. Zeltner, et al., “Near-Perfect Infectivity of Wild-Type AAV as a Benchmark for Infectivity of Recombinant AAV Vectors,” Gene Ther. 17, 872–879 (2010).