By combining the target specificity and long serum half-life of proteins with the potency of small molecules, drug developers can generate products with a distinct therapeutic advantage over either molecule on its own. These bioconjugates are produced through the use of chemical strategies that covalently link a protein or peptide biologic either with a small molecule or another protein or peptide.1
Antibody-drug conjugates (ADCs) are a class of bioconjugates that are the focus of much attention as they show great promise as drug products. However, there are still many opportunities for improving current technologies. For example, the conjugation chemistry used to generate first generation ADCs results in heterogeneous products, which contributes to their complexity. Researchers believe that employing innovative conjugation strategies, instead of more conventional methods, will produce homogeneous products and lead to reduced complexity and increased effectiveness of these drugs. The SMARTag technology, developed by the research group at Catalent (which recently acquired Redwood Bioscience), encompasses novel conjugation chemistries, versatile linker systems, and a modular approach to produce homogeneous best-in-class, site-specifically modified ADCs.
ADCs are a class of bioconjugates that are the focus of much attention as they show great promise as drug products
An ADC comprises three key elements: a monoclonal antibody (mAb), a drug payload (e.g. a cytotoxin), and a linker that connects the mAb to the payload (Figure 1).2 The mAb imparts the ADC with target specificity, directing it to a cell surface antigen that, ideally, has high tumour cell expression and low healthy cell expression. The payload arms the ADC with potency. It kills cells after ADC endocytosis, delivery to the lysosome, and subsequent release into the cytoplasm (Figure 1).
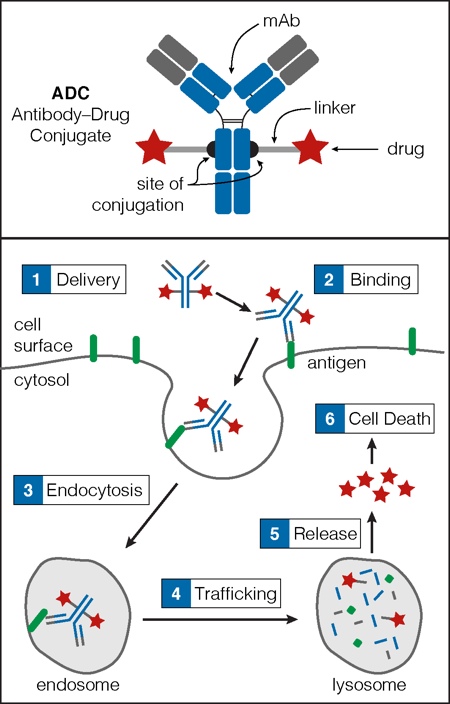
Figure 1: Key components of an ADC
An ADC consists of a mAb, a membrane permeable drug payload (e.g. a toxin) and a chemical linker that connects the mAb to the payload (top). The variable region of the mAb (shown in grey) provides the target specificity as it is the region that binds to the cell surface antigen (bottom). Once the ADC is delivered to the target cell (1), it binds its cell surface antigen (2) and is internalised via endocytosis (3). The ADC then trafficks to the lysosome (4), where lysosomal proteases degrade the ADC into its component parts. The small molecule drug payload is released into the cytoplasm (5) and accesses molecular targets inducing cell death (6).
The linker provides a bridge between the mAb and the pay-load. Before reacting with the mAb, it contains the functional group that participates in conjugation chemistry. Its makeup also affects the overall properties of the payload (e.g. solubility), which in turn can affect the overall properties of the ADC (e.g. potency and stability). The design of an ADC must factor in all three of these key elements to generate a biotherapeutic that will transport a highly potent molecule to the affected target tissue while minimising the off-target effects and patient risk.
A drug’s efficacy relative to its toxicity defines its therapeutic index.3 Conceptually, combining the targeting power of a mAb with the potency of a cytotoxic molecule should produce a drug with a high therapeutic index. Practically, there are significant hurdles to overcome when developing effective, yet safe, ADCs. As a testament to these difficulties, the first ADC was generated more than 30 years ago, yet today, there are only two FDA approved ADCs on the market.
These mixtures of ADCs have been plagued with manufacturing challenges due to batch-to-batch variability and complicated analytics
Historically, the technologies used to build these two approved products result in a heterogeneous milieu of conjugates. These mixtures of ADCs have been plagued with manufacturing challenges due to batch-to-batch variability and complicated analytics. In addition, both efficacy and pharmacokinetic (PK) deficiencies arise from uncontrolled payload number, or drug-to-antibody ratio (DAR). As an alternative, employing site-specific conjugation methods enables homogeneous ADCs that have distinct advantages over conventional ADCs.
Trastuzumab emtansine (Kadcyla) and Brentuximab vedotin (Adcetris), the two ADCs currently on the market, belong to a first generation of ADCs made by conjugating payload-linkers to the mAb through reactive side chains on native amino acid residues (Figure 2).2 Kadcyla is made by conjugating to solvent accessible lysine residues. Given that the underlying mAb, Herceptin, has ~80 lysine residues, precise control of conjugation site is not possible with this conjugation chemistry.
ADCs using lysine conjugation contain molecules with a range of DARs from zero to eight, and can comprise a mixture of up to 70 different conjugated residues (Figure 2).4 In the case of Adcetris, maleimide chemistry is used to conjugate the drug-linker to the free thiols on cysteine residues that are exposed upon partial reduction of the mAb disulfides. There are four disulfides; therefore, depending on the stringency of the reducing conditions, conjugates made using this method can have a DAR of 0, 2, 4, 6 or 8 (Figure 2).5 The optimised method results in conjugates with an average DAR of 4.
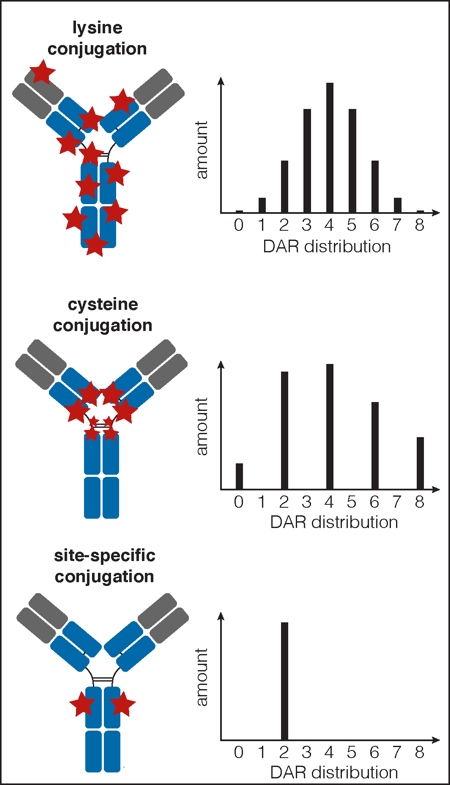
Figure 2: Engineering a site-specific aldehyde into an antibody backbone
The minimal FGE recognition motif/aldehyde tag (CXPXR) is cloned into one or multiple sites within the coding region of the antibody using simple molecular biology techniques. The antibody is produced in an FGE over- expressing cell line, the cysteine within the aldehyde tag is converted by FGE to an aldehyde-bearing formylglycine (fGly), and an antibody containing a reactive aldehyde is produced.
Despite the success of these two ADC products, there are opportunities for improvement. In addition to the heterogeneity associated with conventional ADCs, another drawback is the high drug loading. Seminal work from researchers at Seattle Genetics demonstrated that the stoichiometry of drug loading significantly influences a drug’s PK, efficacy, and toxicity. They found that DAR 4 ADC species were more potent than DAR 2 species but had comparable efficacy and better tolerability than DAR 8 species.
Their conclusion is that ADCs with higher drug loading are more efficacious, yet are cleared from the system faster and have increased toxicity.6 It is therefore important to consider DAR carefully when designing an ADC to achieve the optimal drug loading to maximise therapeutic index. One solution is the SMARTag technology platform, which enables control over DAR through site-specific conjugation methods.
Next generation ADCs
Site-specific ADC technology often involves engineering reactive amino acid residues into the mAb backbone at a specified position (Figure 2). Genentech was the first to introduce a site-specific conjugation method by installing two reactive cysteines at a defined location in the mAb backbone. These cysteines can be conjugated without affecting the native disulfide-bonded cysteine residues to generate ‘THIOMAB’ ADCs with uniform drug loading and a DAR of 2. The THIOMAB technology has yielded ADCs with a higher therapeutic index than conventional ADCs.7,8 In addition, studies show that conjugating to differential sites on the mAb leads to drastically different efficacy and PK profiles.9 These results suggest that homogeneity is not enough; careful consideration must also be given to the site of conjugation.
There are several ADC technologies that not only provide site-specificity, but also offer the flexibility to engineer a conjugation site across different regions of a mAb
There are several ADC technologies, including THIOMAB, that not only provide site-specificity, but also offer the flexibility to engineer a conjugation site across different regions of a mAb. These methods can produce homogeneous products with a DAR of 2 with one engineered site per heavy or light chain monomer (or a higher DAR with more engineered sites). Ambrx, Sutro and Allozyne engineer conjugation sites into the mAb by incorporating non-natural amino acids containing bio-orthogonal reactive groups.10,11
Pfizer/Rinat incorporates a LLQG tag into mAbs, which serves as a substrate for transglutaminase catalysed amidation.12 Catalent employs a novel chemoenzymatic approach, using the naturally occurring formylglycine-generating enzyme (FGE), to introduce an aldehyde tag for site-specific conjugation (Figure 3).13 Studies from many of these groups have demonstrated that site of conjugation can have a dramatic impact on in vitro and in vivo properties of the ADC.12,14,15
Exploring structure-activity relationships (SAR): Catalent is pioneering the generation of optimised therapeutics by the assembly of ADCs using a SAR approach. The design of ADC therapeutics, with defined molecular architecture (bioconjugate SAR), is accomplished using the SMARTag technology platform. This includes the full suite of ADC components: flexible payload site selection, novel and versatile aldehyde conjugation chemistry, stable, modular and adaptable triggers for payload release, and a diverse library of payloads (Figure 4).
This approach to making ADCs begins with the simple engineering of a short FGE recognition site (CXPXR, the ‘aldehyde tag’) into any mAb or protein of interest at a desired location on the heavy or light chain. The aldehyde-tagged mAb is then expressed in an FGE overexpressing mammalian cell line (e.g. the Catalent’s proprietary GPEx cell lines). During protein expression, FGE recognises the cysteine residue within the consensus sequence and co-translationally converts it to formylglycine (fGly), generating high-titer mAbs with two fGly residues (and therefore two uniquely reactive aldehyde groups) per mAb molecule (Figure 3).13 After purification, the mAb is ready for bioorthogonal conjugation chemistry at one or more specific sites of interest.
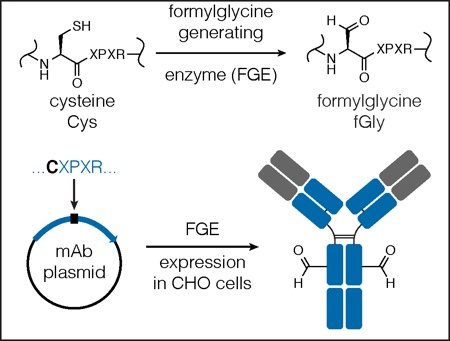
Figure 3: Drug distribution and DAR profiles for different ADC conjugation methods
Conjugation to lysine or cysteine residues results in a wide distribution of DAR, whereas site-specific conjugation results in a specified DAR (depending on the number of engineered sites)
Historically, aldehydes were conjugated with aminooxy or hydrazine nucleophiles to yield oxime or hydrazide products. Although performed under aqueous conditions, these reactions are fairly slow, take place under acidic conditions, and are susceptible to hydrolysis, undermining their usefulness when long-term stability is required (i.e. with ADCs). Catalent developed an aldehyde-specific conjugation chemistry, known as the Hydrazino-iso-Pictet-Spengler (HIPS) reaction, which can be performed near neutral pH, is faster than aminooxy ligations, and results in conjugates with long-term stability.15,16 The HIPS reaction exploits the speed of alpha affect nucleophiles, to generate a reactive intermediate, which closes shut to form a stable C-C bond between the mAb and payload.
To survey SAR in ADC design, Catalent generated a panel of site-selective ADCs using HIPS chemistry. These molecules contained a maytansine cytotoxic drug-linker, conjugated at various positions along the heavy chain and light chain of an α-Her2 mAb. The goal was to determine if placement of the aldehyde tag (and thus of the maytansine) had an impact on in vitro and/or in vivo behaviour. The site-specific ADCs were benchmarked to α-Her2-DM1, which is made with the same toxin and mAb as Catalent ADCs but uses conventional lysine conjugation chemistry and has twice the drug loading.
Catalent is pioneering the generation of optimised therapeutics by the assembly of ADCs using a structure-activity relationships approach
Independent of tag site, SMARTag ADCs showed excellent in vitro cytotoxicity, with IC50s in the low picomolar range, similar to free maytansine and α-Her2-DM1. These ADCs were tested in an NCI-N87 xenograft model, where they all showed antitumour activity. However, in contrast to the in vitro results, the efficacy of the ADC depended on the placement of the payload. PK studies in mice revealed that these differences in efficacy correlate with differences in serum half-lives. The SMARTag ADC tagged at the C-terminus of the heavy chain was the most efficacious and exhibited a greater half-life than the other SMARTag ADCs and α-Her2-DM1.15
These were quintessential SAR studies where differences in activity were attributed to slight structural differences in the ADC, which may be caused by changes in the local chemical and structural environment surrounding the tag site. Catalent believes that, using its technology, there are opportunities to optimise ADCs by further exploring the SAR of different tag placements. The platform also affords the opportunity to explore SAR surrounding payload and linker composition. HIPS conjugation chemistry is compatible with different classes of payloads and toxins with different modes of action, including tubulin disruptors and DNA alkylators (Figure 4). In addition, HIPS chemistry is compatible with both cleavable and non-cleavable linker systems and a variety of stable linker components, adding great versatility to the platform (Figure 4).
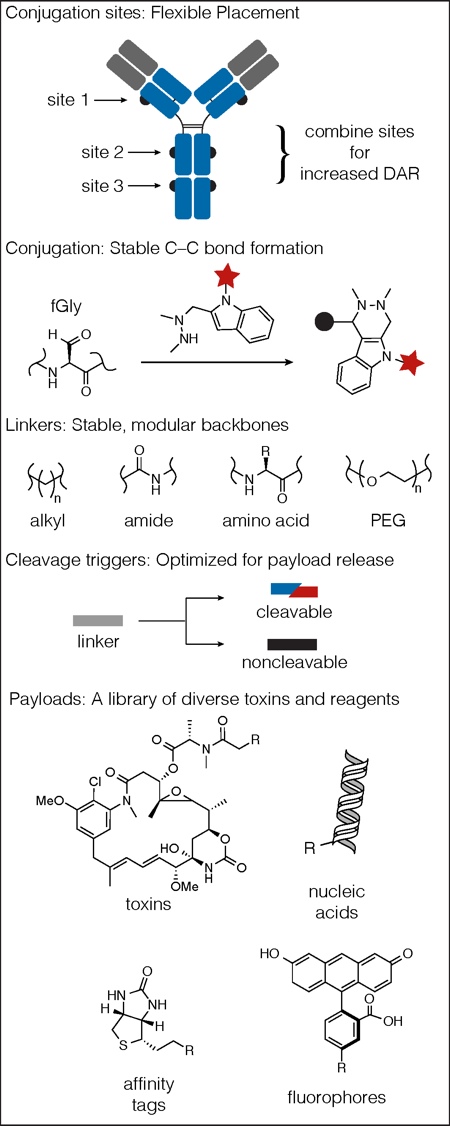
Figure 4: The SMARTag technology platform encompasses flexible placement of conjugation sites, stable HIPS chemistry, and a library of linkers, cleavage triggers and payloads.
Catalent generated a panel of C-terminally aldehyde-tagged α-Her2-maytansine ADCs that varied only in the non-cleavable linker portion and then examined the effect of linker composition on the conjugation efficiency and in vitro and in vivo properties of the SMARTag ADCs. It was found that solubility of the linker is very important for efficiency of the conjugation reaction. Incorporation of PEG spacers conferred some solubility but it was the addition of amino acid residues that imparted enough solubility to ensure efficient conjugation to fGly. Interestingly, among the soluble linkers tested in vivo, it was found that very minor changes to the linker caused drastic differences in efficacy that could not have been predicted based on in vitro cytotoxicity results.17 These data highlight the fact that linker design can have a major impact on in vivo outcomes.
Based on the results from the tag placement and linker SAR studies, an exploratory rat toxicology study was performed with an ADC containing an optimal linker conjugated to an optimal tag site. This ADC, containing a noncleavable maytansine payload, was well-tolerated in rats. It caused no mortality even at a previously observed lethal dose when using a conventional α-Her2-DM1.15 Many hypothesise that a more homogeneous drug product, with controlled DAR and drug placement, will have a greater therapeutic index due to less toxicity and perhaps greater efficacy. These data lend some credence to this hypothesis. Given the tolerance of Catalent’s site-specific DAR 2 ADC, the company is now exploring higher DAR site-specific ADCs. Lower expressing targets may be better treated with higher DAR species and provide access to a greater number of therapeutic targets.
The SMARTag platform incorporates the three major components of an ADC (mAb, the payload, linker). Each piece must be carefully considered when designing an ADC to enhance its overall in vitro and in vivo properties. Site-specific conjugation chemistry yields homogeneous and stable ADCs that exhibit strong antitumour effects, long half-lives, and reduced toxicity compared with conventional ADCs, suggesting an increased therapeutic index. With more than 30 ADCs currently in clinical trials and many site-specific ADCs in pre-clinical development, the future of targeted drug delivery is poised to realise its potential.
The authors
Robyn M. Barfield is Group Leader, Bioconjugation, Patrick G. Holder, Research Scientist and David Rabuka Global Head of R&D, Chemical Biology, all at Catalent Pharma Solutions.
References
1. N. Stephanopoulos, M. B. Francis. Nature Chemical Biol. 7 (12) 2011: 876–884
2. E. L. Sievers, P. D. Senter. Annu. Rev. Med. 64, 2013:15–29.
3. P. Y. Muller, M. N. Milton. Nat. Rev. Drug Discovery 11, 2012: 751–761.
4. Wang, et al. Protein Sci, 14(9), 2005: 2436–2446.
5. M. M. C. Sun et al. Bioconjugate Chem. 16(5) 2005: 1282–1290.
6. K. J. Hamblett et al. Clin. Cancer Res. 10(20) 2004: 7063–7070.
7. J. R. Junutula, et al. Nat. Biotechnol. 26(8) 2008: 928–932.
8. S. C. Jeffrey et al. Bioconj. Chem. 24(7) 2013: 1256–1263.
9. B. Shen et al. Nat. Biotechnol. 30(2) 2012: 184–191.
10. J. Y. Axup et al. Proc. Natl. Acad. Sci. 109(40) 2012: 16101–16106.
11. E. S. Zimmerman et al. Bioconj. Chem. 25(2) 2014: 351–361.
12. P. Strop et al. Chem. Biol. 20(2) 2013: 161–167.
13. D. Rabuka et al. Nat. Protocols 7(6) 2012: 1052–1067.
14. F. Tian et al. Proc. Natl. Acad. Sci. 111(5) 2014: 1766–1771.
15. P. M. Drake et al. Bioconj. Chem. 25(7) 2014: 1331–1341.
16. P. Agarwal et al. Bioconjug Chem 24(6) 2013 :846–51.
17. A. E. Albers et al. Eur. J. Med. Chem. 88 2014: 3–9.
Kadcyla is a registered trademark of Genentech, Inc. Adcetris is a registered trademark of Seattle Genetics, Inc.






