The use of virtual screening in lead identification has seen much refinement since its inception, Paul Finn, ceo of InhibOx in Oxford, UK, reviews it current use and advantages as well as future challenges
The pharmaceutical industry is facing a productivity crisis. This is surprising, since industry expenditure on R&D has grown rapidly to a total this year of $60 billion; investments have been made in a plethora of new technologies; and the Human Genome Project, together with an explosion in our understanding of the mechanistic origins of diseases from neurodegeneration to cancer, has led to a dramatic increase in the number of drug discovery targets. Despite this, output, as measured by new drug approvals, is static or even declining.
One underlying cause can perhaps be found in the nature of the novel targets that genomics is uncovering. Historically, the industry has worked on surprisingly few targets, estimated in one study to be only about 400. Furthermore, these targets are drawn from very few protein families. G-protein coupled receptors (GPCR), ion channels and nuclear hormone receptors and some enzymes are heavily represented in the list of drug targets, other gene families are hardly represented at all.
Over the last decade, high-throughput screening (HTS) has been the predominant technology used to identify new leads. It has been, is, and will continue to be, a good source of leads. However, no technology is without its disadvantages, and HTS has limitations in respect of new targets. The likelihood of identifying leads from HTS depends (amongst other things) on the quantity and quality of the compound screening collection. The size of HTS screening collections ranges from a few million in large pharma companies to perhaps a few tens of thousands in biotech. Compared with the universe of possible drug molecules, these numbers are tiny.
Estimates of the size of the druglike chemical universe involve many approximations and uncertainties, but even the most conservative estimates place it at billions of times larger than even pharma company collections. In addition, these collections are highly biased to particular regions of chemistry space - companies have made many similar molecules, for example aromatic amines as part of their GPCR programmes.
There is also the pragmatic focus towards more tractable syntheses (Fig. 1). The cost of building large scale HTS is already too great for any but the largest players, and to increase the size of the existing collections by orders of magnitude is not only prohibitively expensive, but also presents enormous practical issues.
The issue is perhaps even more pressing for the biotech sector. Typically such companies have novel biology, perhaps a new target identified from academic research. There may be valuable IP in the target itself, but ultimately, long-term value will come from the small molecule inhibitors or antagonists that are developed against the target. Unfortunately, the costs of progressing from target biology to lead identification and beyond are significant, and biotech companies are often very resource limited, and so cannot afford a major investment in HTS technology.
In summary, the genomic revolution has shifted a bottleneck in drug discovery from target identification to lead identification. However, experimental approaches (primarily HTS) have severe limitations in overcoming this bottleneck. If experimental solutions to the lead identification bottleneck are limited, can computational approaches provide a way forward?
Computer-aided drug design
The idea of using computers to assist the drug discovery process is not new. As early as the 1960s statistical techniques were being used to help understand structure-activity relationships and guide lead optimization. In the 1980s and 1990s, with the growth of structural biology new computational techniques were added to the armamentarium (structure-based design), again with a focus on lead optimisation.
More recently, methods have been developed which impact on lead identification. Two basic approaches have been adopted. In the first - de novo design - the algorithm builds a molecule (usually many molecules) atom by atom, or group by group in the active site of a protein to have a high predicted affinity. The most attractive molecules, usually as judged by human experts, are then selected for synthesis and testing. These molecules are almost guaranteed to be novel, but a drawback of the approach is that the molecules designed are often difficult to synthesize, and this issue has limited adoption of the approach, although progress continues to be made. In the second approach, which has been much more widely adopted and which is generically called virtual screening, a database of molecules is searched for compounds which meet certain activity criteria.
Virtual screening
Virtual screening is a computational technique that is being increasingly adopted by the industry to complement HTS. In outline the process is simple and closely analogous to its experimental counterpart (Fig.2). In HTS, the key components are a miniaturised assay, a compound collection, and the physical systems (robotics and informatics) to perform the tests. Each of these has a computational equivalent.
Instead of an experimental assay, virtual screening has a computational model of activity. Different models are possible, but they are in general, three-dimensional - a typical example would be a geometrical arrangement of chemical functionality that defines a minimum requirement for activity. This will often be derived from the protein structure of the target, but could also come from an analysis of a set of known ligands if the target structure is unknown.
Instead of a physical compound collection, one has a computer database of molecules. This database can be much larger than any existing physical collection. It can also, by design, be much more chemically diverse.
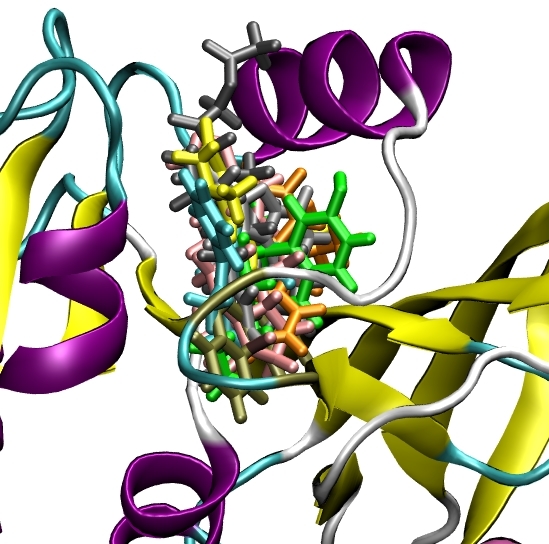
The final component is a computer algorithm that can assess each molecule in the database for its ability to satisfy the activity model. Many different algorithms have been developed, and this is a highly active area of current research. The usual objective is to identify a very small subset of the database for experimental testing, typically 100-200 compounds. An example result is shown in Figure 3 - there are many ways of occupying the binding site, using varied structural frameworks.
screening advantages
A virtual screening approach to lead identification can address some of the key issues highlighted above. Molecular databases can be much larger than their experimental counterparts. Typical virtual screening databases, such as the one at InhibOx, have several components: a collection of several million commercially available molecules.
A major advantage of these compounds is that they are readily available and inexpensive. This allows a rapid validation of the virtual screening model and generation of some initial SAR information. Compared with running a full HTS campaign this can be very time- and cost-effective and may allow a company with more limited resources to exploit some biological knowledge and assets that otherwise would not be possible.
Another component is combinatorial libraries, based on known chemistry and available starting materials (from the commercial part of the database). These libraries can either be generic, or constructed specifically with a particular target in mind. Whilst this requires greater resource input than screening available compounds, the advantage is the molecules are much more likely to be proprietary, especially if this is built around in-house chemistry expertise. This is also a natural second step after first obtaining encouraging results from commercial molecule screening.
Again, making a small targeted array of compounds, rather than a fully combinatorial library, is an attractive proposition. Such approaches also offer options for chemistry providers to add value to their products in a market that is becoming increasingly price sensitive due to the pressures created by the emerging markets.
Virtual screening can also offer advantages to companies that have HTS capability. HTS capacity may be limited, a particular molecular target may not be compatible with HTS, HTS may have failed to identify a suitable lead, or a speculative target may not be judged sufficiently validated or interesting to warrant the expenditure of a full HTS campaign, but be worth the much smaller resource required for the virtual screening approach
Given these advantages, it is not surprising that we are seeing a rapid growth in interest in virtual screening. Figure 4 shows the growth in virtual screening articles, covering both methodological developments and application examples. An example of the scale that is possible is given by the Cancer Screensaver project. This worldwide, distributed computing project applied virtual screening to a number of cancer targets. In excess of 2.5 million people volunteered their computer time, and a database of 3.5 billion molecules was screened. Whilst this is a public interest rather than an industrial application, it illustrates what is possible with current technology.
future challenges
If all this is true, then why has virtual screening not taken over from HTS, and solved the lead identification crisis already? Virtual screening is a technology still in its infancy. Many algorithms have been developed, and these are successful by many measures. For example, in benchmarking experiments many virtual screening methods can successfully reproduce experimental crystal structure data to better than 2A RMS (the accepted measure of a 'good' fit) about 70-80% of the time. Figure 5 shows a representative example from our own work.
However, when compounds predicted to be active by virtual screening are tested experimentally, a very large false positive rate is observed. In other words many of the predictions are incorrect, and a hit rate of only a few percent is considered to be a good result. Whilst this is enough to be of practical utility (a 5% hit rate would identify 10 actives from a typical virtual screening selection of 200 compounds), it is clearly far from ideal.
Molecular interactions are complicated - both ligand and protein have complex shapes, are flexible, and are interacting in an aqueous environment. Quantum mechanics and molecular dynamics are capable in many cases of correctly calculating the strength of these interactions, but they are very computationally intensive. Therefore, to enable the large-scale searching performed in virtual screening, simplifications must be made to the description, and these simplifications introduce errors.
A major focus of our work, and of others, is to develop new methods that combine the accuracy of rigorous methods with scale. This may well require more than the incremental improvement of existing approaches, but a more radical recasting of the framework in which molecular interactions are described, drawing on work from other areas of application, in particular solid state physics and computer science. If this research is successful, then, perhaps, the pharmaceutical industry can join other technology led industries such as aerospace in the widespread use of simulation prior to expensive experimental implementation.






