It’s widely accepted that the new European Union (EU) regulation for medical devices will have major implications for the labelling operations of every manufacturer that trades in the EU. EU MDR, which began a 5-year preimplementation period in May 2017, will be fully adopted in 2022. However, the first major milestone — the introduction of an EU Database for Medical Devices (EUDAMED) — is on track to launch in 2020. Many of the labelling challenges wrapped up in the regulation will need to be ironed out by then. The clock has been ticking for a year.
The labelling of medical devices is already complex, encountering multiple organisational touchpoints across a diverse end-to-end process. It is why labelling has become a mission-critical business system for medical device companies. However, EU MDR brings added complexity and is forcing companies to review their labelling infrastructure as they battle for organisational preparedness. It’s a battle that won’t be won overnight — but it needs to start now.

The definition of ‘label’ has, in the past decade, expanded to include items such as multi language booklets and Instructions for Use (IFUs). EU MDR introduces new rules that pertain to these crucial materials. But what does it mean for the sticky label itself? EU MDR introduces additional information that needs to be included on labels, forcing organisations to design new label templates that make room for data not previously part of the labelling system. It’s both a design and a data challenge … and they must quickly be addressed to avoid a sticky situation.
The main headline is emphatic: every manufacturer that ships products to the EU will have to change their labels. Every, single, one. The regulation requires that all labels must include a standardised symbol to indicate that a package contains a medical device. Although the specific symbol has yet to be determined, it’s unlikely that adding it to the label will pose problems for manufacturers. However, other requirements present more complex challenges. Many companies will need to rethink their label designs — and the systems they use to support them — if they’re to be compliant by 2020. Individual requirements are outlined in section 23.2 of the regulation. Though not all of them affect every manufacturer, a number have significant implications. Certainly, every change that requires new information to be added to the label will have operational repercussions.
This is particularly the case for products that already have busy labels or where the size of devices leaves little ‘real estate’ to work with. Here are some of the most impactful changes, beginning with those with implications for everyone.
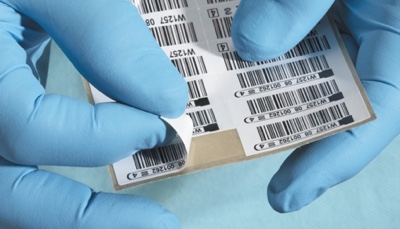
UDI applied in Europe: UDI (unique device identifier) has its own dedicated section within EU MDR. The EU’s guidelines are in line with the Global UDI initiative, as are the FDA UDI rules; all labels must include PI (GTIN) and DI components as textual and barcoded content. Many companies have already solved the UDI challenge because of its requirement in the US. However, companies who do not ship to the US are at the beginning of the UDI journey.
More serial and lot numbers: This change falls under the UDI remit. However, EU MDR requires more products to be serialised than FDA UDI. Every active implantable device must have its own unique serial number. Other implantable devices will require a serial or lot number. Making the shift from batch labelling to a world in which individual products need to be married with the right label at the right time is challenging. It requires a data-led labelling system.
Highlight authorised EU representatives: Every manufacturer whose registered place of business is outside the EU is required to have a licensed EU representative. Previously, details of that representative were included in the IFU. EU MDR requires companies to print that information — symbol, name and address — on the label.
Warnings and precautions must be on-label: This change will probably have the biggest impact. EU MDR mandates that all warnings relating to a device must be printed on the label. Previously, these were all included in the IFU. Although the regulation says information can be kept to a minimum — with more detail in the IFU — compliance will require companies to add ‘written text’ to labels, bringing the issue of translation into play. It will also lead to variability in the space such phrases consume, with some languages requiring more room than others. The choice of which warnings need to be included is left to the manufacturer. However, the criteria on which they should base those decisions remain unclear.
Label must indicate blood and tissue derivatives: EU MDR provides regulation for medical technology innovations not previously covered by MDD; that is, nanotechnology, the use of computer software or medicines. All devices that incorporate a medical substance or tissues/cells or their derivatives must clearly indicate this on the label.
Label must indicate CMR or endocrine-disrupting substance: Devices with a presence of carcinogenic, mutagenic or toxic to reproduction (CMR) substances must declare so on the label. These requirements go much further than MDD.
Include reprocessing cycles: This is another huge data challenge. Under EU MDR, labels for single-use devices that can be reprocessed must detail the maximum amount a device can be reprocessed as well as the number of times the individual device has been reprocessed to date. Manufacturers will need to integrate batch information from their ERP systems and identify data changes to the product definition. This requires capturing information that’s not currently included in the labelling system. Some manufacturers are considering stopping reprocessing single-use devices altogether — a major strategic decision that illustrates the potential impact of EU MDR.
eIFUs (electronic Information for Use): EU MDR also introduces requirements around electronic IFUs and the ‘absorption of substances’ that dictate changes in labelling processes. Manufacturers must clearly indicate that the instructions for use are supplied in electronic rather than in paper form. This information must be provided on the packaging for each unit and — in the case of fixed medical devices — on the device itself. Manufacturers must also provide information regarding how to access the instructions for use in electronic form, such as through the addition of a URL.
Medical device symbol: Under EU MDR, manufacturers of medical devices must now include a new field on their labels — a clear symbolic indication that the device is a medical device. Again, this requirement goes much further than MDD and further impacts the design, spacing and data inputs to medical device labels.
Label spacing differences: To address the fact that new mandatory content — such as the addition of warnings and precautions — is almost certainly going to create congestion, the recent final ruling by FDA no longer requires English text to appear alongside symbols to free up label space. This will be worth consideration as its impact will lead to potentially significant label redesign.
Gluing it all together
Achieving compliance with EU MDR will naturally create labelling challenges for medical device companies. Companies therefore need to ensure their current labelling system is fit for purpose. But they need to do it soon. Although EU MDR will not be fully enforced until 2022, the birth of EUDAMED in 2020 means companies could be prevented from registering or reregistering products if they don’t address associated labelling challenges ahead of its introduction. Failure to do so could mean that companies can’t market their products in Europe.
The most effective labelling systems will be those that give manufactures 360° visibility of all their master data assets — and that provide the core components of label lifecycle management to ensure critical data from across the global organisation is glued together whenever — and wherever — it’s required. Moreover, they will provide a flexibility that enables manufacturers to respond to future changes in regulations.
Although UDI and EU MDR have naturally focused thinking, companies should be mindful that regulatory change is imminent in other important markets. With emerging markets now key drivers of growth, companies must ensure that their labelling system is future-proofed to adapt to change in all the regions where they trade. Otherwise, like all good labels, they might just end up stuck.
This article appeared in the September issue of Manufacturing Chemist.