Traditionally, selecting an enabling technology consists of a trial-and-error approach to find the best technology for a specific molecule, which consumes both time and resources. Computational tools that model the movement and interactions of atoms and molecules can be effective in helping to advance molecules more rapidly to the clinic and to market.
Pharma and biotech innovators may use innovations in molecular dynamics simulation (MDS) to reduce the timelines, complexity and risk associated with complex molecules.
Unlocking computational power in formulation development
We define a computational tool as a platform or a device that leverages the analytical capacity of a computer via a human:computer/smart device interface to predict, interpret or inform a particular outcome that is ordinarily experimentally measured. Molecular dynamics are computer simulations that model the movement and interactions of atoms and molecules with time.
From the trajectory of each of the atoms and molecules within the simulation, equilibrium and dynamic experimental properties can be extracted. At Lonza, we have been creating an MDS platform specifically designed to support the formulation development process.
Our initial focus areas for applying MDS include
Selection of an appropriate enabling technology for poorly water-soluble drugs: informed technology selection, incorporating quality-by-design principles, based on drug structure alone following drug discovery. Concurrent trial-and-error technology exploration is avoided and development time reduced.
Food-effect risk assessment: anticipation of a food-effect using drug structure prior to preclinical/clinical testing to reduce programme risk and timelines.
Design, development and molecular analysis of drug containing lipid-based formulations (LBFs) in the anhydrous, dispersed and digested states: excipient screening and rank order selection, concept LBF design, colloidal fate of drug as the LBF moves along the GI tract and overall LBF performance risk evaluation is defined.
Before we could apply MDS to key areas of formulation development, we worked to develop proprietary computational methods and molecular forcefields (sets of equations and parameters that model intramolecular and interatomic interactions) that are applicable and accurately model drugs and commonly used lipidic excipients.
This is in addition to the endogenous compounds in the gut that perform key interactions with drugs and formulations, such as bile salts and phospholipids. For example, Figure 1 shows the improvement in modelling of the dispersed behaviour of a non-ionic surfactant molecule using a previously developed MDS forcefield for protein research (53a6) compared with our first-generation MDS forcefield for lipidic excipients (53a6DBW).
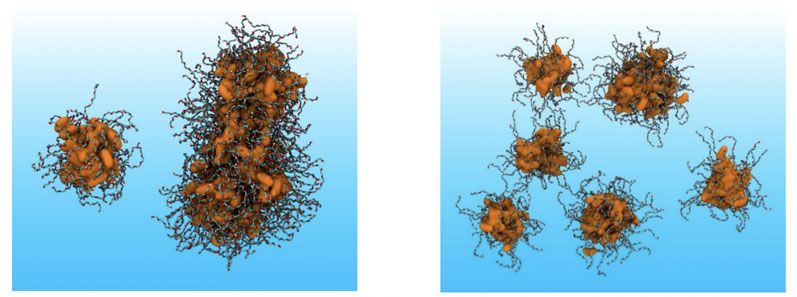
Figure 1: Aggregation behaviour of PEG-8 monocaprylate in water with the given force fields; left = 53a6 and right = 53a6DBW. The latter correctly predicts dispersion behaviour of the surfactant. Key: alkane, hydrophobic core represented with a solvent accessible surface area and PEG chains with liquorice representation. The atomic colouring is as follows: cyan = CH2 and CH3, white = polar hydrogens, red = oxygen and orange = alkane CH2 and CH3, with water omitted for clarity.
Our MDS computational methods and forcefield development are continually being refined and we are currently working on a second-generation MDS forcefield to yield even more accurate results.
Our MDS development has been complemented by rigorous validation using experimental data, which is an essential part of development for any computational tool. Some example validation data that we have collected include
- the correlation of MDS predicted and experimentally determined logP values1
- predicted colloidal phase of the dispersed non-ionic surfactant Kolliphor EL2
- diffusion coefficients of bile salt/phospholipid colloids3
- the phase behaviour of lipidic excipients4
- the position of drug molecules of varying lipophilicity in surfactant micelles or in oil-rich formulations dispersed in water.5,6
As a result of this work and the optimisation of our MDS methods and forcefields, we are now capable and confident in modelling complex and dynamic lipidic systems, including peptide formulations designed for oral delivery.
The cyclic peptide cyclosporine A was initially formulated as an LBF by Sandoz AG (later becoming Novartis) to improve oral bioavailability of the peptide and was approved by the FDA in 1990 under the Sandimmune brand.
The Sandimmune formulation showed significant patient variability and a positive food-effect, which was attributed to it forming a coarse emulsion in water and dependence on lipid digestion to promote absorption.7
A new LBF was strategically developed by Novartis to show improved dispersibility and, in turn, improved pharmacokinetic properties. This LBF was Neoral, approved in 1995 and remains the gold-standard formulation to deliver cyclosporine A.
To explore the effectiveness of MDS, we performed a reflective analysis of Sandimmune and Neoral formulations using our platform, first confirming the much improved dispersibility of Neoral versus Sandimmune (Figure 2A/B) and then the dramatic increase in colloidal dispersion of cyclosporine when the Sandimmune formulation is digested.
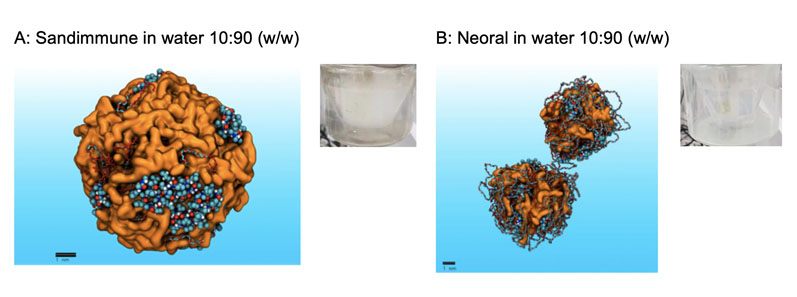
Figure 2: Showing MDS models of the dispersion behaviour of Sandimmune and Neoral in water. MDS correctly predicts the agglomeration of Sandimmune (A), consistent with experimental observations (milky dispersion) and the superior dispersion of Neoral (B), consistent with fine dispersion observed experimentally (clear dispersion).
The significant change in size and number of aggregates in fasted state simulated intestinal fluids or FaSSIF (Figure 3A Left) compared with fed state simulated intestinal fluids or FeSSIF (Figure 3A Right) is notable, indicating a formulation food-effect.
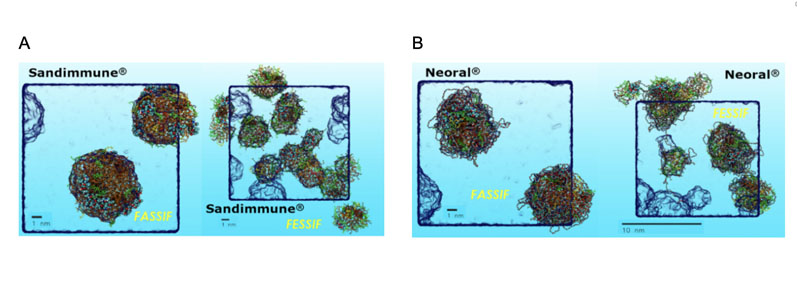
Figure 3: Showing the MDS predictions of Sandimmune (A) and Neoral (B) under simulated fasted (FaSSIF) and fed (FeSSIF) conditions (left and right, respectively), highlighting effect of formulation digestion specifically the greater difference in performance for Sandimmune. Key: cyan = carbon, red = oxygen, white = polar hydrogen,PEG surfactants are straws in cyan/red, glycerides and alkane chains in orange, bile salts in green and phospholipids in yellow.
This case study highlights how computational development tools such as MDS can be applied to realise the rapid identification of concept formulations for small molecule and peptide drugs.
The power of MDS versus other modelling platforms
MDS provides the potential of 24/7 computational power to formulation development. With access to computational tools that are tailored to formulation development comes the possibility to change the way formulation development is conducted, with formulators combining laboratory and computational experiments to achieve a robust performing and stable formulation that can be progressed into the clinic.
Those interested in the field should be aware of the difference in computational power of MDS versus other molecular modelling platforms, such as Chem3D (PerkinElmer Informatics) and Maestro (Schrödiger LLC) as these only have the capacity to model single molecules in a vacuum and not the interactions between drugs, formulations and the surrounding environment.
For comparison, our typical MDS runs can involve calculating the position of more than 300,000 atoms (drug, formulation, water, bile salts and phospholipids, etc.) during a short period of time (usually around 100 ns).
Furthermore, there are different types of MDS, such as coarse-grain or united-atom methods. There are pros and cons of both techniques.
Coarse-grain MDS enables the analysis of larger systems without significantly increasing the CPU time burden; but, to model large systems, it is necessary to simplify a collection of atoms into a single “cluster,” meaning that significant detail at the molecular level is lost, compromising the overall value of the MDS approach.
We utilise united-atom MDS to probe molecular level interactions. However, to generate reliable data, united-atom MDS requires highly accurate simulations and no compromise in detail.
Current capabilities and looking ahead
As computational power increases exponentially related to Moore’s law, so does the ability to model larger and more complex systems that better approach reality.8
Whereas only small molecules in a vacuum could be modelled for a limited length of time, modern computational tools are approaching the ability to accurately model complex biological systems in real-time.
For example, the complexity of the influenza A H1N1 2009 virus, consisting of 160 million atoms spanning 115 nm in diameter, has implications on the detail available and the questions that can be asked by simulations.9
The complexity allows for the exciting prospect of probing key interactions at the molecular level in such systems.
Molecular complexity is important, particularly in the area of therapeutic peptide development wherein the size of the drug can quickly become computationally burdensome. For example, semaglutide contains more than 580 atoms per molecule, each with bond angles and energies to be calculated simultaneously, as well as key interactions with solvent and other proximal molecules.
At Lonza, we are currently capable of running routine simulations of systems containing more than 300,000 molecules within several days, utilising a high-performance computing (HPC) cluster. This capability allows us to model significantly more complex lipid formulation systems.
Importantly for drug development, MDS provides an alternative to historical trial-and-error approaches. We can now use MDS for the informed selection of enabling technology at an early development stage to accelerate the development of peptides targeting oral delivery.
This prevents matching target compound with the wrong technology or trialling multiple technologies in parallel, both at a cost of time and resources. Additionally, MDS allows formulations to be screened in silico before precious API and formulator resources are wasted, which can thereby improve OH&S conditions owing to reduced exposure to potentially toxic compounds.
One of the greatest means to reduce development timelines is formulation assessment and derisking. The molecular information garnered from MDS allows drug developers to look for potential vulnerabilities of peptides in formulations under stressing conditions.
Understanding the behaviour of the API within a given formulation and the formulation itself within a specific solution (water, FaSSIF or FeSSIF) helps to derisk formulation approaches for APIs as well as aid in identifying concept formulations — all of which contribute to reducing development time.
Additionally, as mentioned earlier, 24/7 screening of formulations utilising MDS can significantly reduce development timelines. Most importantly, development using MDS can start as soon as the chemical structure is provided, whereas the traditional approach would require waiting for shipping of compounds, internal risk assessment and several other time-costing hurdles.
Summary
Continued innovation is critical to successfully formulate and advance complex molecules. MDS models can aid in the rapid development of bioavailability challenged molecules, including helping to design lipid-based formulations that are appropriate for peptide molecules.
As the drug development pipeline continues to consist of more and more low-bioavailability molecules, computational tools may be a powerful implement for drug developers to advance candidate molecules. Contract development and manufacturing organisations (CDMOs) can use MDS tools to help biopharma companies enhance molecule bioavailability and rapidly move new medicines forward into clinical trials and to market.
References
- D.B. Warren, et al., "Improvement in the Predicted Partitioning of Alcohol and Polyethylene Oxide Groups Between Water and Octanol (logP) in Molecular Dynamics Simulations," J. Pharm. Sci. 108(1), 214–222 (2019).
- E.J.A. Suys, et al., "A Nonionic Polyethylene Oxide (PEO) Surfactant Model: Experimental and Molecular Dynamics Studies of Kolliphor EL," J. Pharm. Sci. 108(1), 193–204 (2019).
- W.A. Birru, et al., "Computational Models of the Gastrointestinal Environment. 1. The Effect of Digestion on the Phase Behavior of Intestinal Fluids," Mol. Pharm. 14(3), 566–579 (2017).
- W.A. Birru, et al., "Digestion of Phospholipids After Secretion of Bile into the Duodenum Changes the Phase Behavior of Bile Components," Mol. Pharm. 11(8), 2825–2834 (2014).
- D.B. Warren, et al., "Location of Solvated Probe Molecules within Nonionic Surfactant Micelles Using Molecular Dynamics," J. Pharm. Sci. 108(1), 205–213 (2019).
- D.B. Warren, et al., "Glyceride Lipid Formulations: Molecular Dynamics Modeling of Phase Behavior During Dispersion and Molecular Interactions Between Drugs and Excipients," Pharm. Res. 30(12), 3238–3253 (2013).
- E.A. Mueller, et al., "Influence of a Fat-Rich Meal on the Pharmacokinetics of a New Oral Formulation of Cyclosporine in a Crossover Comparison with the Market Formulation," Pharm. Res. 11(1), 151–155 (1994).
- G.E. Moore, "Cramming More Components onto Integrated Circuits," Electronics 38(8), 4–8 (1965).
- J.D. Durrant, et al., "Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism," ACS Cent. Sci. 6(2), 189–196 (2020).
The authors wish to thank Dr Dallas Warren (Research Fellow, Monash University) for fruitful discussions.
